

Hereditary spastic paraplegias: identification of a novel SPG57 variant affecting TFG oligomerization and description of HSP subtypes in Sudan
- PMID: 27601211
- PMCID: PMC5159756
- DOI: 10.1038/ejhg.2016.108
Hereditary spastic paraplegias: identification of a novel SPG57 variant affecting TFG oligomerization and description of HSP subtypes in Sudan
Abstract
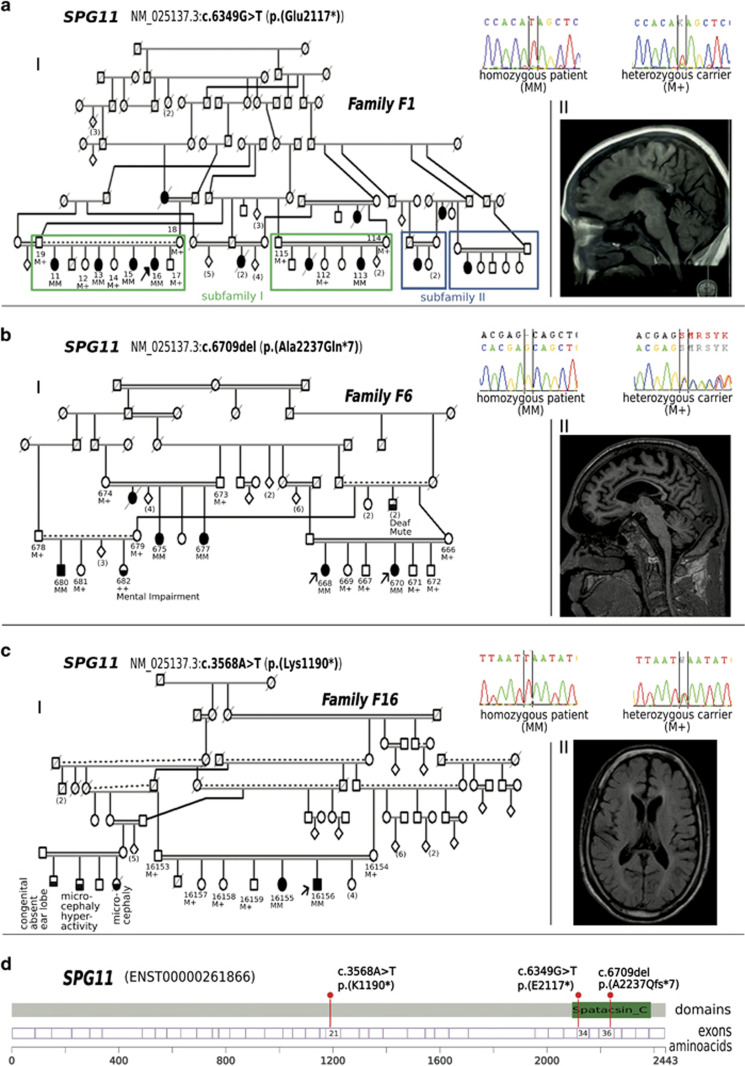
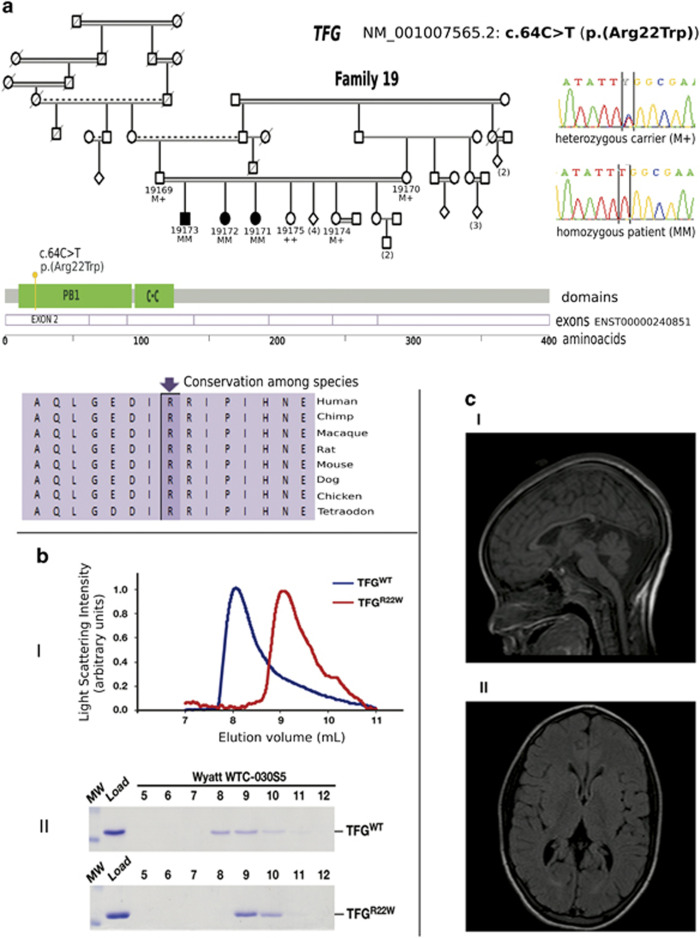
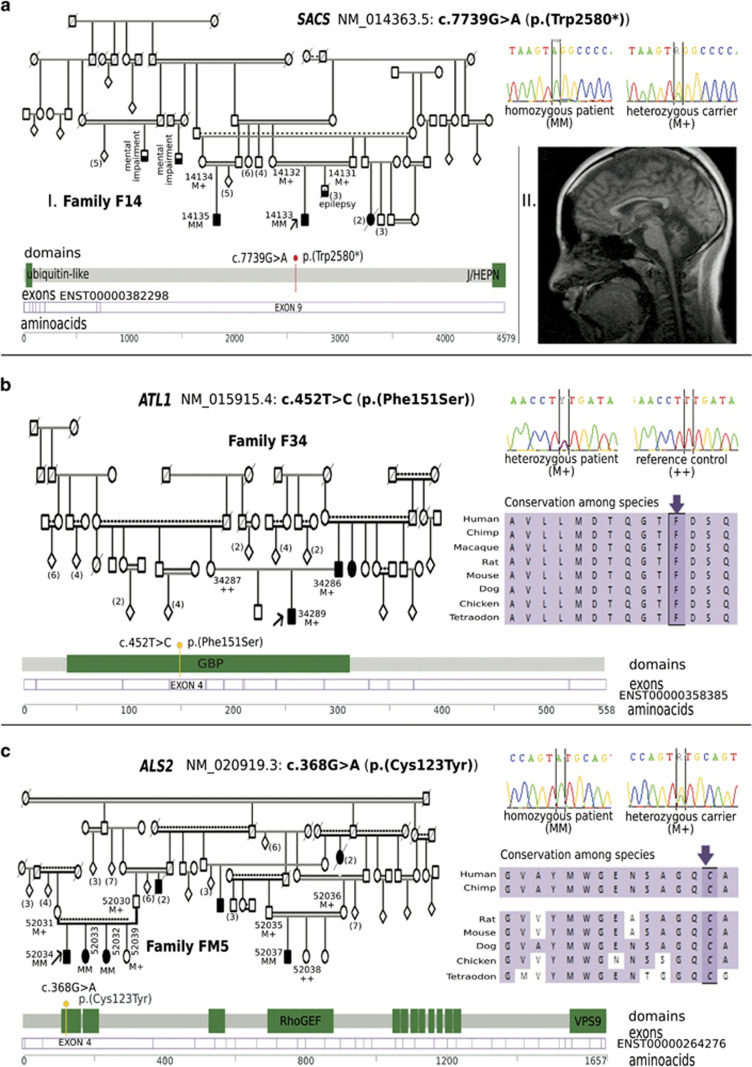
Hereditary spastic paraplegias (HSP) are the second most common type of motor neuron disease recognized worldwide. We investigated a total of 25 consanguineous families from Sudan. We used next-generation sequencing to screen 74 HSP-related genes in 23 families. Linkage analysis and candidate gene sequencing was performed in two other families. We established a genetic diagnosis in six families with autosomal recessive HSP (SPG11 in three families and TFG/SPG57, SACS and ALS2 in one family each). A heterozygous mutation in a gene involved in an autosomal dominant HSP (ATL1/SPG3A) was also identified in one additional family. Six out of seven identified variants were novel. The c.64C>T (p.(Arg22Trp)) TFG/SPG57 variant (PB1 domain) is the second identified that underlies HSP, and we demonstrated its impact on TFG oligomerization in vitro. Patients did not present with visual impairment as observed in a previously reported SPG57 family (c.316C>T (p.(Arg106Cys)) in coiled-coil domain), suggesting unique contributions of the PB1 and coiled-coil domains in TFG complex formation/function and a possible phenotype correlation to variant location. Some families manifested marked phenotypic variations implying the possibility of modifier factors complicated by high inbreeding. Finally, additional genetic heterogeneity is expected in HSP Sudanese families. The remaining families might unravel new genes or uncommon modes of inheritance.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Fink J: Hereditary spastic paraplegia overview. In: Pagon R, Adam M, Ardinger H et al: (eds): GeneReviews. University of Washington: Seattle (WA), USA, 1993. Available at: http://www.ncbi.nlm.nih.gov/books/NBK1509/. - PubMed
-
- Schmitz-Hübsch T, Klockgether T: An update on inherited ataxias. Curr Neurol Neurosci Rep 2008; 8: 310–319. - PubMed
-
- Finsterer J: Ataxias with autosomal, X-chromosomal or maternal inheritance. Can J Neurol Sci 2009; 36: 409–428. - PubMed
-
- Coutelier M, Stevanin G, Brice A: Genetic landscape remodelling in spinocerebellar ataxias: the influence of next-generation sequencing. J Neurol 2015; 262: 2382–2395. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
