

Genome editing using CRISPR-Cas9 to create the HPFH genotype in HSPCs: An approach for treating sickle cell disease and β-thalassemia
- PMID: 27601644
- PMCID: PMC5035856
- DOI: 10.1073/pnas.1612075113
Genome editing using CRISPR-Cas9 to create the HPFH genotype in HSPCs: An approach for treating sickle cell disease and β-thalassemia
Abstract
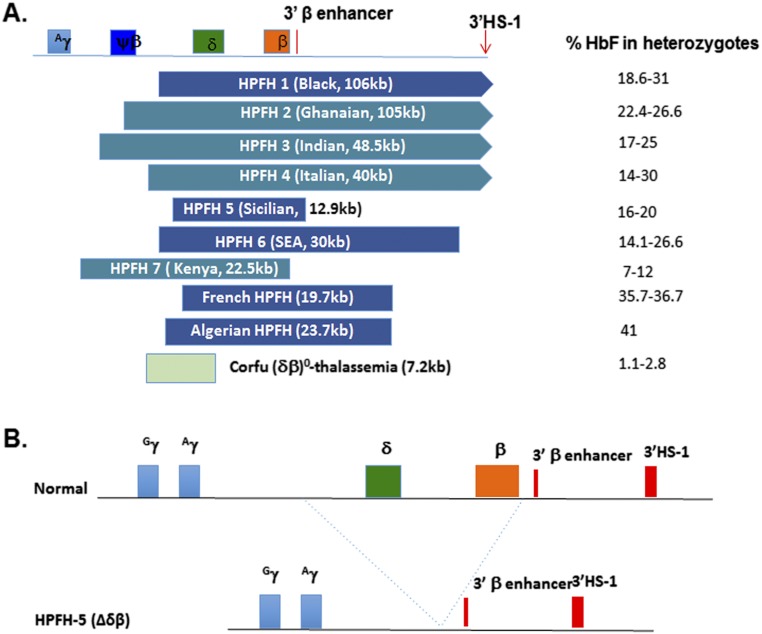
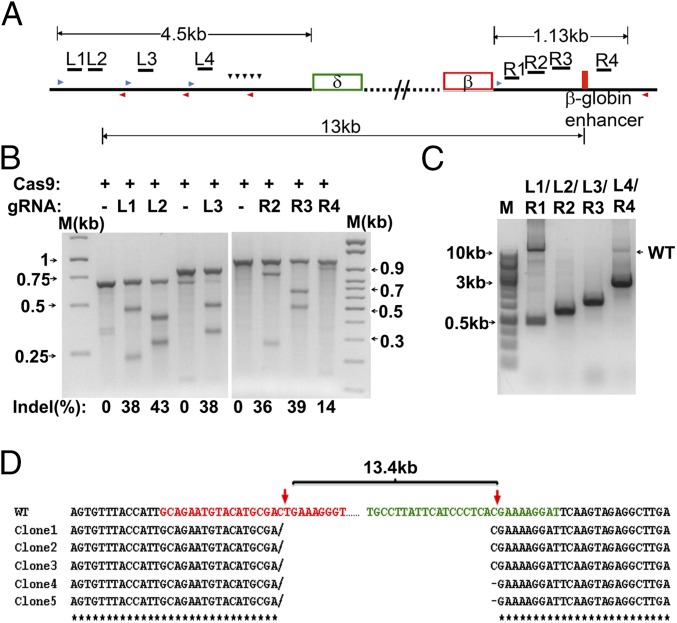
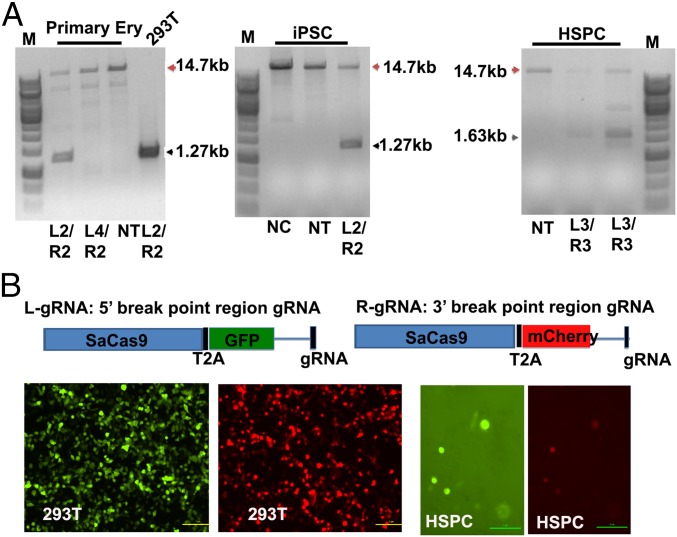
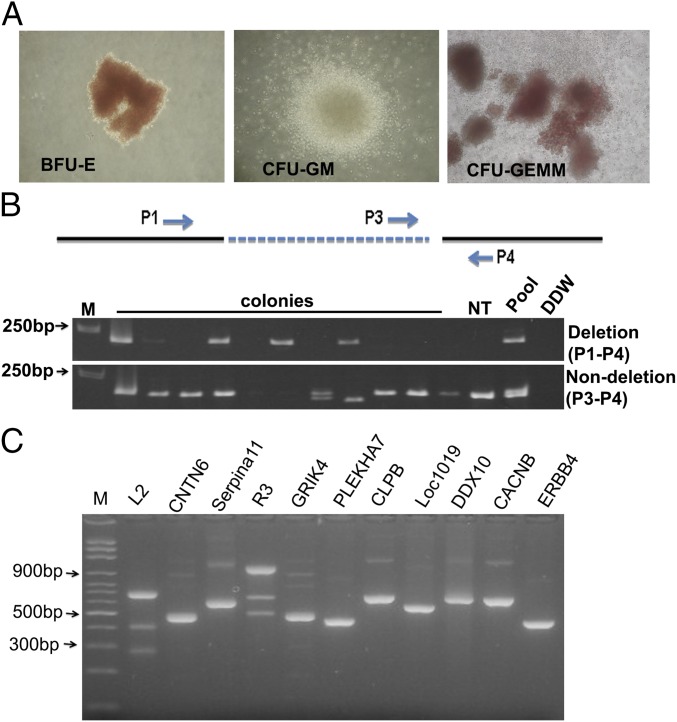
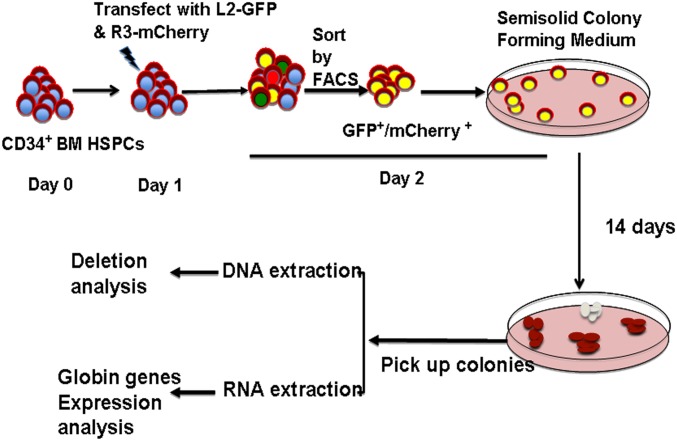
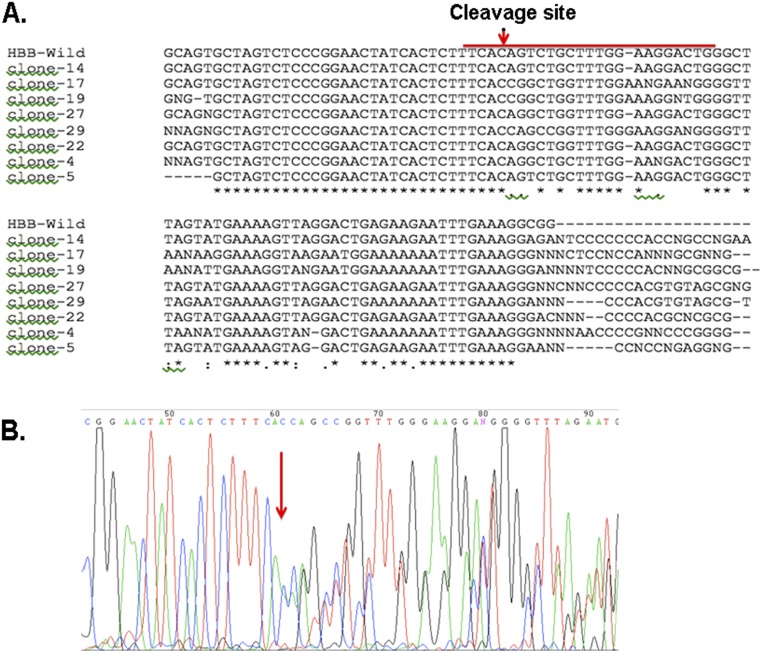
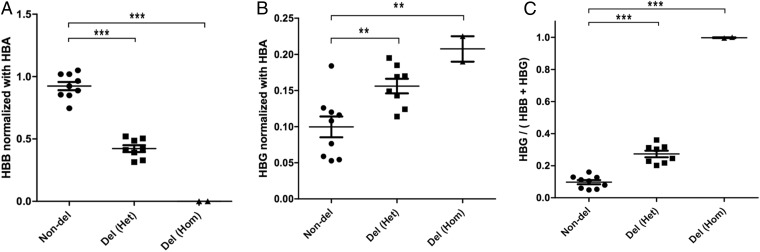
Hereditary persistence of fetal hemoglobin (HPFH) is a condition in some individuals who have a high level of fetal hemoglobin throughout life. Individuals with compound heterozygous β-thalassemia or sickle cell disease (SCD) and HPFH have milder clinical manifestations. Using RNA-guided clustered regularly interspaced short palindromic repeats-associated Cas9 (CRISPR-Cas9) genome-editing technology, we deleted, in normal hematopoietic stem and progenitor cells (HSPCs), 13 kb of the β-globin locus to mimic the naturally occurring Sicilian HPFH mutation. The efficiency of targeting deletion reached 31% in cells with the delivery of both upstream and downstream breakpoint guide RNA (gRNA)-guided Staphylococcus aureus Cas9 nuclease (SaCas9). The erythroid colonies differentiated from HSPCs with HPFH deletion showed significantly higher γ-globin gene expression compared with the colonies without deletion. By T7 endonuclease 1 assay, we did not detect any off-target effects in the colonies with deletion. We propose that this strategy of using nonhomologous end joining (NHEJ) to modify the genome may provide an efficient approach toward the development of a safe autologous transplantation for patients with homozygous β-thalassemia and SCD.
Keywords: colony assay; deletion; engineered nucleases; erythroid differentiation; fetal hemoglobin.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Bank A, Dorazio R, Leboulch P. A phase I/II clinical trial of beta-globin gene therapy for beta-thalassemia. Ann N Y Acad Sci. 2005;1054:308–316. - PubMed
-
- Persons DA. Hematopoietic stem cell gene transfer for the treatment of hemoglobin disorders. Hematology Am Soc Hematol Educ Program. 2009;2009:690–697. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
