

Therapeutic targeting of splicing in cancer
- PMID: 27603132
- PMCID: PMC5644489
- DOI: 10.1038/nm.4165
Therapeutic targeting of splicing in cancer
Abstract
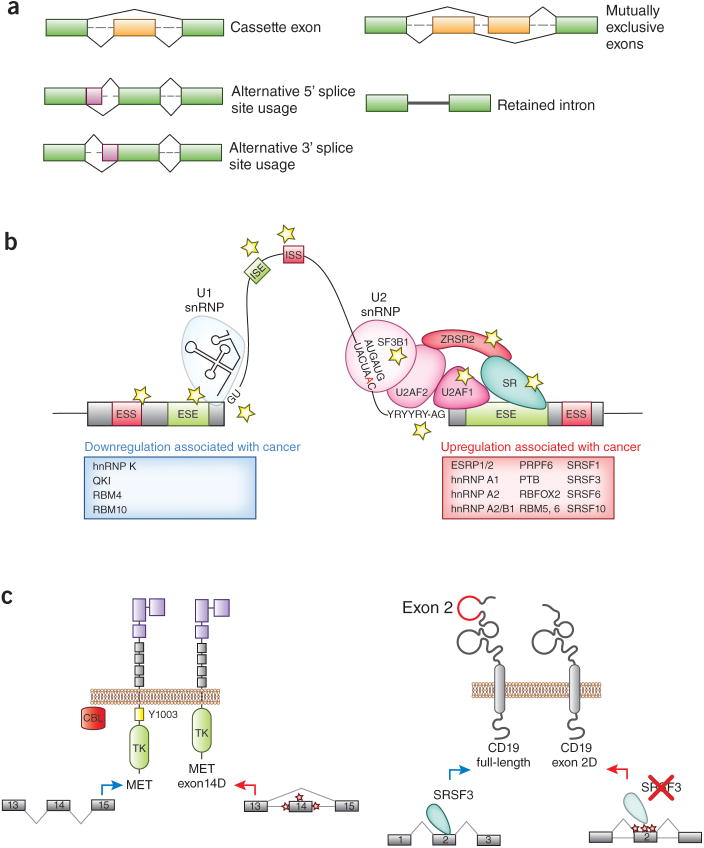
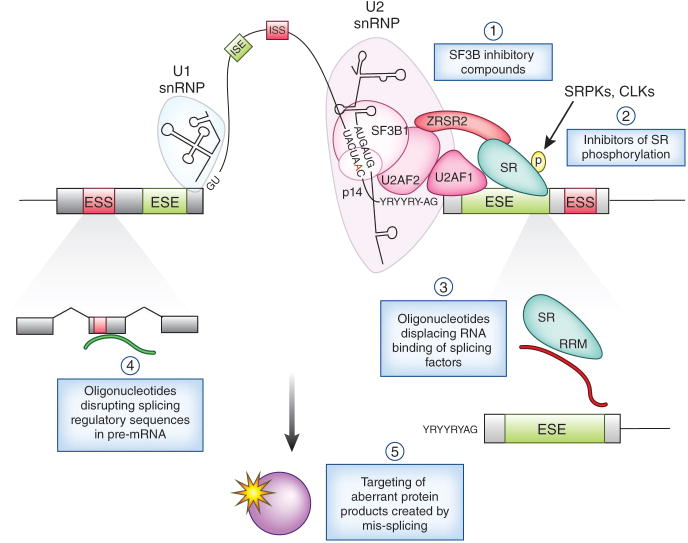
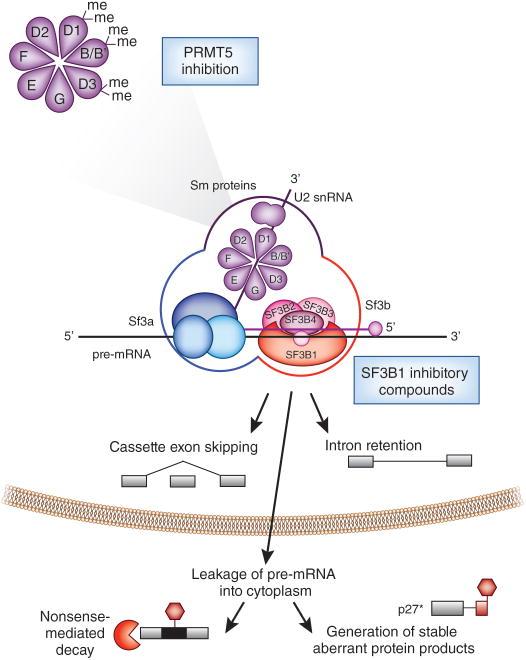
Recent studies have highlighted that splicing patterns are frequently altered in cancer and that mutations in genes encoding spliceosomal proteins, as well as mutations affecting the splicing of key cancer-associated genes, are enriched in cancer. In parallel, there is also accumulating evidence that several molecular subtypes of cancer are highly dependent on splicing function for cell survival. These findings have resulted in a growing interest in targeting splicing catalysis, splicing regulatory proteins, and/or specific key altered splicing events in the treatment of cancer. Here we present strategies that exist and that are in development to target altered dependency on the spliceosome, as well as aberrant splicing, in cancer. These include drugs to target global splicing in cancer subtypes that are preferentially dependent on wild-type splicing for survival, methods to alter post-translational modifications of splicing-regulating proteins, and strategies to modulate pathologic splicing events and protein-RNA interactions in cancer.
Figures
References
-
- Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nature genetics. 2008;40:1413–1415. - PubMed
-
- Supek F, Miñana B, Valcárcel J, Gabaldón T, Lehner B. Synonymous mutations frequently act as driver mutations in human cancers. Cell. 2014;156:1324–1335. - PubMed
-
- Jung H, et al. Intron retention is a widespread mechanism of tumor-suppressor inactivation. Nature genetics. 2015;47:1242–1248. - PubMed
-
- Yoshida K, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64–69. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
