

Demonstration of Protein-Based Human Identification Using the Hair Shaft Proteome
- PMID: 27603779
- PMCID: PMC5014411
- DOI: 10.1371/journal.pone.0160653
Demonstration of Protein-Based Human Identification Using the Hair Shaft Proteome
Abstract
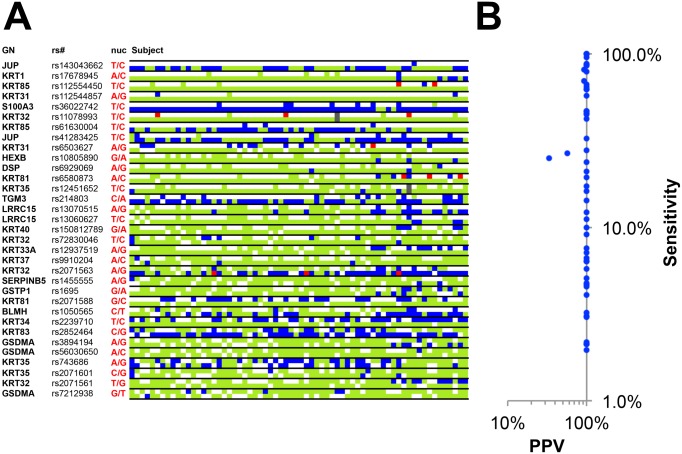
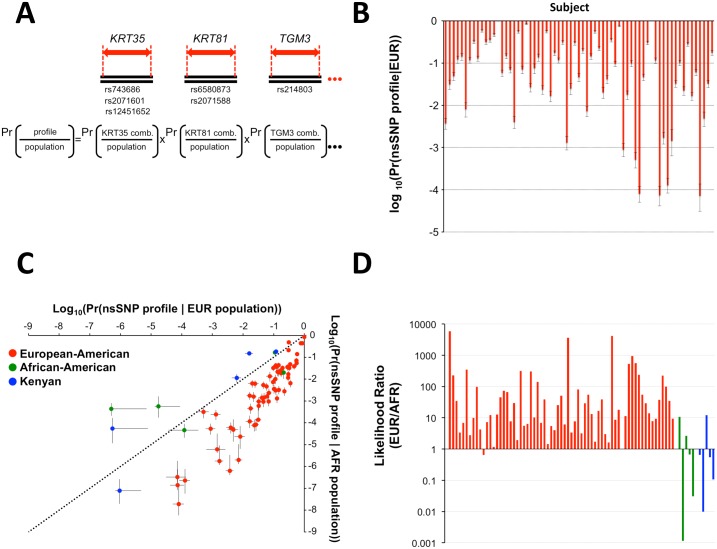
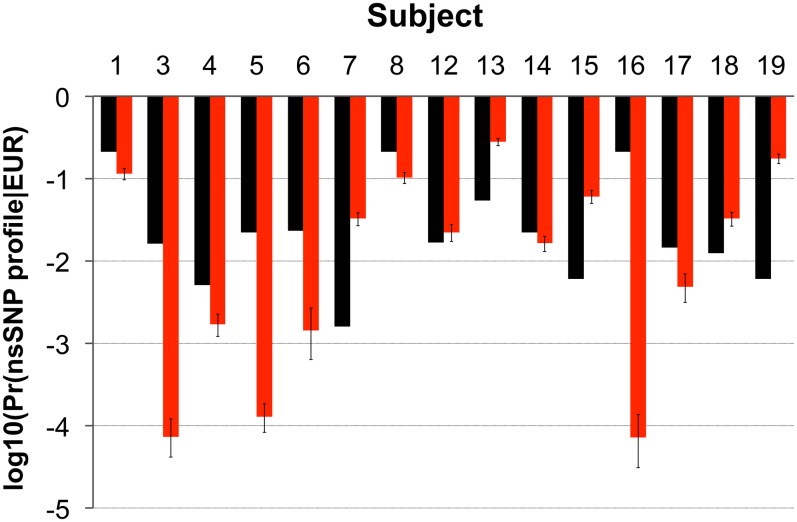
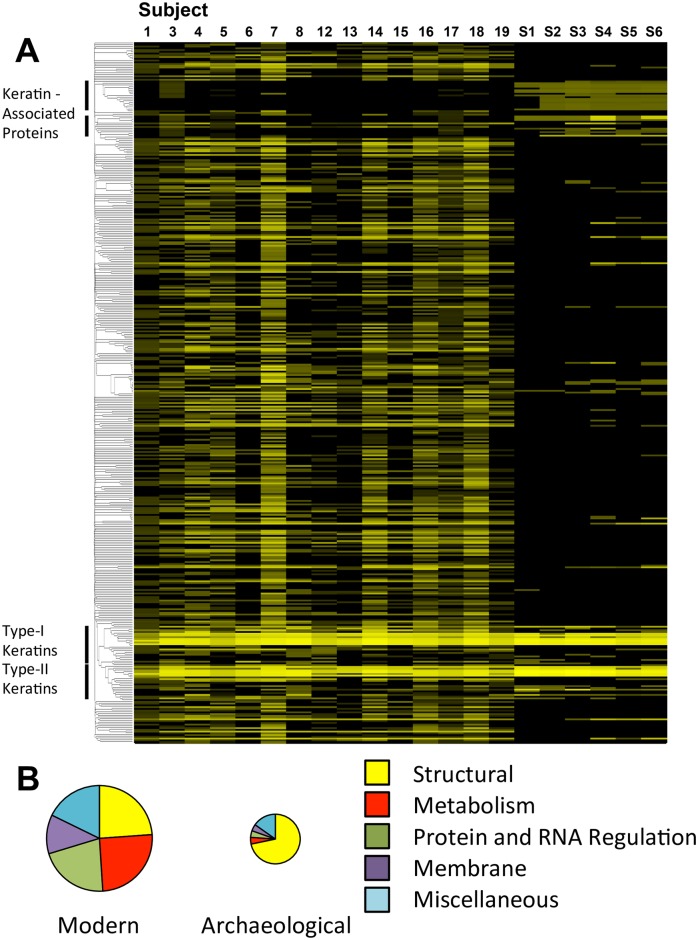
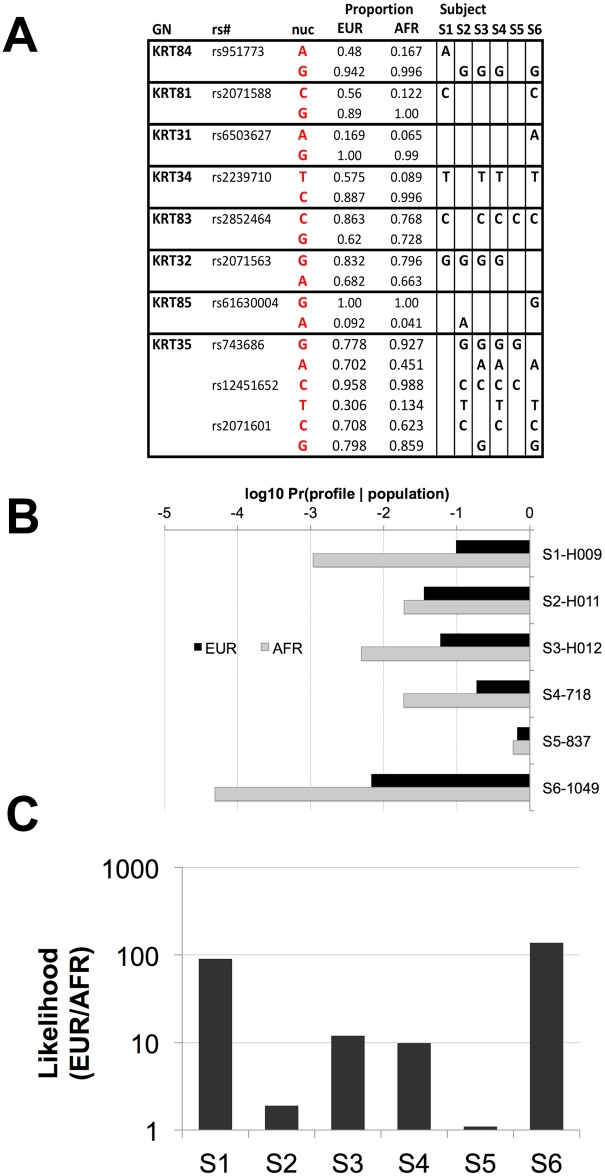
Human identification from biological material is largely dependent on the ability to characterize genetic polymorphisms in DNA. Unfortunately, DNA can degrade in the environment, sometimes below the level at which it can be amplified by PCR. Protein however is chemically more robust than DNA and can persist for longer periods. Protein also contains genetic variation in the form of single amino acid polymorphisms. These can be used to infer the status of non-synonymous single nucleotide polymorphism alleles. To demonstrate this, we used mass spectrometry-based shotgun proteomics to characterize hair shaft proteins in 66 European-American subjects. A total of 596 single nucleotide polymorphism alleles were correctly imputed in 32 loci from 22 genes of subjects' DNA and directly validated using Sanger sequencing. Estimates of the probability of resulting individual non-synonymous single nucleotide polymorphism allelic profiles in the European population, using the product rule, resulted in a maximum power of discrimination of 1 in 12,500. Imputed non-synonymous single nucleotide polymorphism profiles from European-American subjects were considerably less frequent in the African population (maximum likelihood ratio = 11,000). The converse was true for hair shafts collected from an additional 10 subjects with African ancestry, where some profiles were more frequent in the African population. Genetically variant peptides were also identified in hair shaft datasets from six archaeological skeletal remains (up to 260 years old). This study demonstrates that quantifiable measures of identity discrimination and biogeographic background can be obtained from detecting genetically variant peptides in hair shaft protein, including hair from bioarchaeological contexts.
Conflict of interest statement
Patent based on the concept and some data presented in this study have been awarded (US 8,877,455 B2, Australian Patent 2011229918, Canadian Patent CA 2794248, and European Patent EP11759843.3, GJP inventor). The patent is owned by Parker Proteomics LLC. Protein-Based Identification Technologies LLC has an exclusive license to develop the intellectual property and is co-owned by Utah Valley University and GJP. This ownership of PBIT and associated intellectual property does not alter our adherence to PLOS ONE policies on sharing data and materials.
Figures
References
-
- The National Research Council. Strengthening Forensic Science in the United States: A Path Forward Washington D.C.: The National Academy Press; 2009. September 9, 2009.
-
- Butler JM. Fundamentals of Forensic DNA Typing: Academic Press; 2010.
MeSH terms
Grants and funding
- UC2 HL103010/HL/NHLBI NIH HHS/United States
- RC2 HL102926/HL/NHLBI NIH HHS/United States
- P42 ES004699/ES/NIEHS NIH HHS/United States
- RC2 HL102924/HL/NHLBI NIH HHS/United States
- UL1 TR000002/TR/NCATS NIH HHS/United States
- RC2 HL103010/HL/NHLBI NIH HHS/United States
- P20 RR024237/RR/NCRR NIH HHS/United States
- RC2 HL102923/HL/NHLBI NIH HHS/United States
- UC2 HL102926/HL/NHLBI NIH HHS/United States
- UC2 HL102923/HL/NHLBI NIH HHS/United States
- UC2 HL102924/HL/NHLBI NIH HHS/United States
- UL1 TR001067/TR/NCATS NIH HHS/United States
- P20 RR020185/RR/NCRR NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
