

Evolutionary Dynamics of Abundant Stop Codon Readthrough
- PMID: 27604222
- PMCID: PMC5100048
- DOI: 10.1093/molbev/msw189
Evolutionary Dynamics of Abundant Stop Codon Readthrough
Abstract
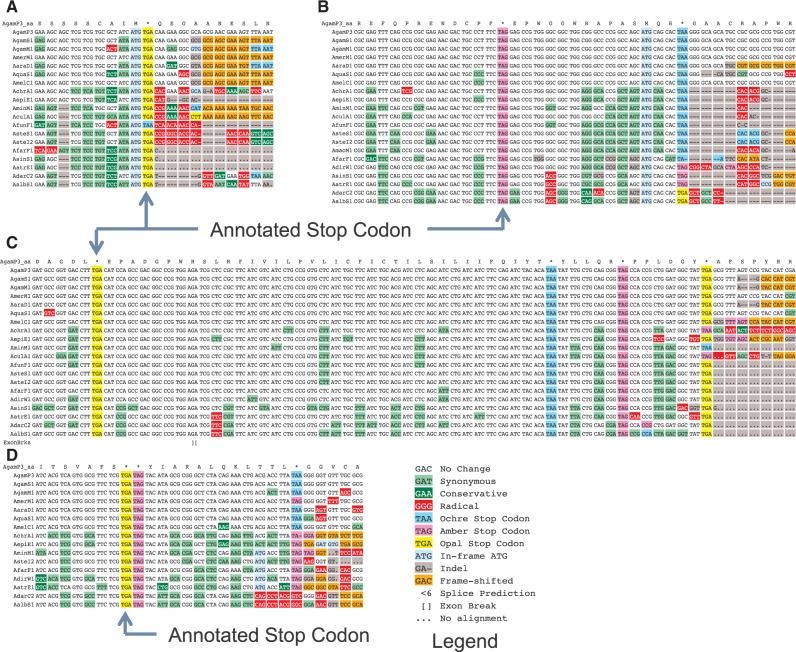
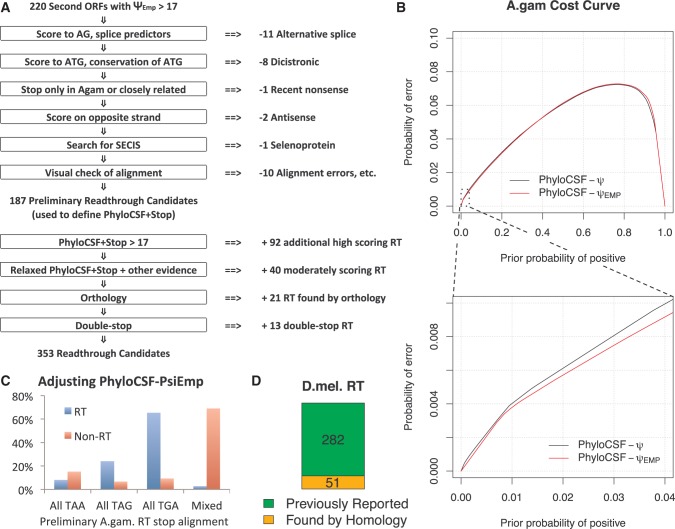
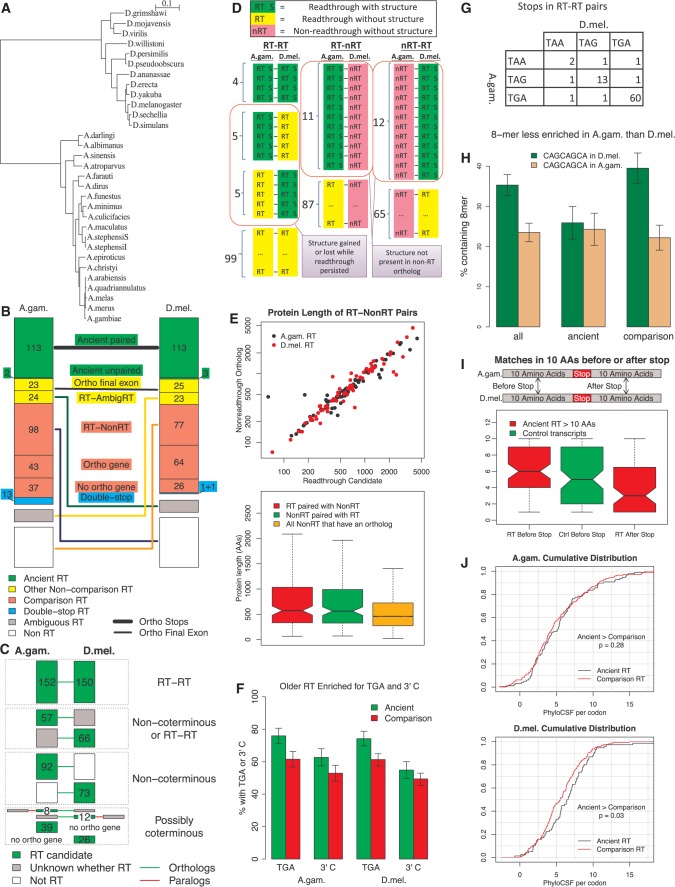
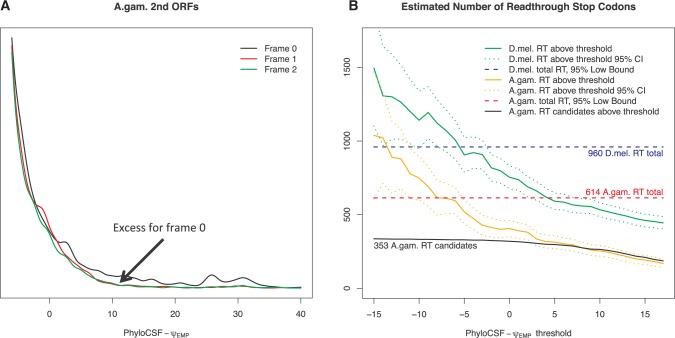
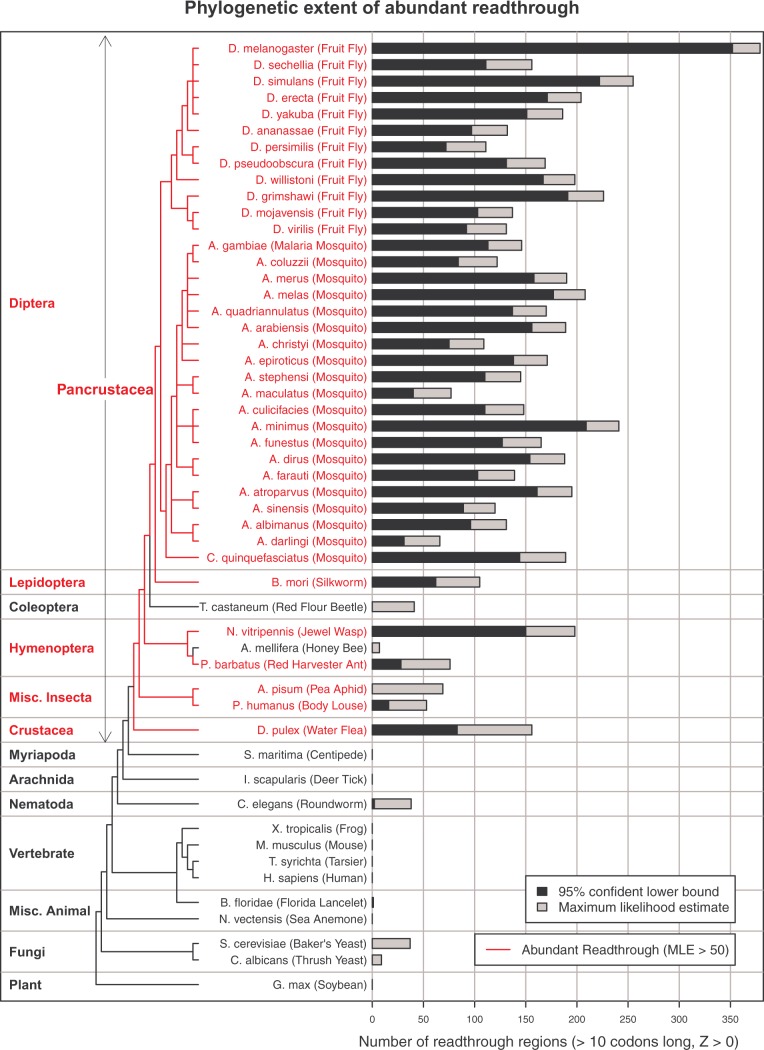
Translational stop codon readthrough emerged as a major regulatory mechanism affecting hundreds of genes in animal genomes, based on recent comparative genomics and ribosomal profiling evidence, but its evolutionary properties remain unknown. Here, we leverage comparative genomic evidence across 21 Anopheles mosquitoes to systematically annotate readthrough genes in the malaria vector Anopheles gambiae, and to provide the first study of abundant readthrough evolution, by comparison with 20 Drosophila species. Using improved comparative genomics methods for detecting readthrough, we identify evolutionary signatures of conserved, functional readthrough of 353 stop codons in the malaria vector, Anopheles gambiae, and of 51 additional Drosophila melanogaster stop codons, including several cases of double and triple readthrough and of readthrough of two adjacent stop codons. We find that most differences between the readthrough repertoires of the two species arose from readthrough gain or loss in existing genes, rather than birth of new genes or gene death; that readthrough-associated RNA structures are sometimes gained or lost while readthrough persists; that readthrough is more likely to be lost at TAA and TAG stop codons; and that readthrough is under continued purifying evolutionary selection in mosquito, based on population genetic evidence. We also determine readthrough-associated gene properties that predate readthrough, and identify differences in the characteristic properties of readthrough genes between clades. We estimate more than 600 functional readthrough stop codons in mosquito and 900 in fruit fly, provide evidence of readthrough control of peroxisomal targeting, and refine the phylogenetic extent of abundant readthrough as following divergence from centipede.
Keywords: Anopheles; Drosophila; recoding.; stop codon readthrough; termination codon suppression; translational readthrough.
© The Author 2016. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures
References
-
- Baudin-Baillieu A, Legendre R, Kuchly C, Hatin I, Demais S, Mestdagh C, Gautheret D, Namy O. 2014. Genome-wide translational changes induced by the prion [PSI+]. Cell Rep. 8:439–448. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
