

An expanded evaluation of protein function prediction methods shows an improvement in accuracy
- PMID: 27604469
- PMCID: PMC5015320
- DOI: 10.1186/s13059-016-1037-6
An expanded evaluation of protein function prediction methods shows an improvement in accuracy
Abstract
Background: A major bottleneck in our understanding of the molecular underpinnings of life is the assignment of function to proteins. While molecular experiments provide the most reliable annotation of proteins, their relatively low throughput and restricted purview have led to an increasing role for computational function prediction. However, assessing methods for protein function prediction and tracking progress in the field remain challenging.
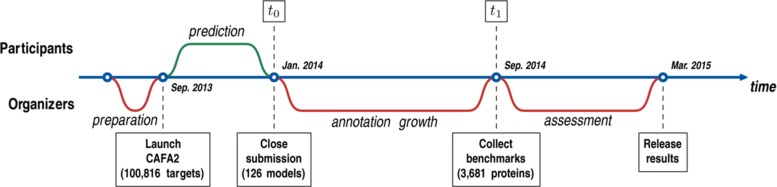
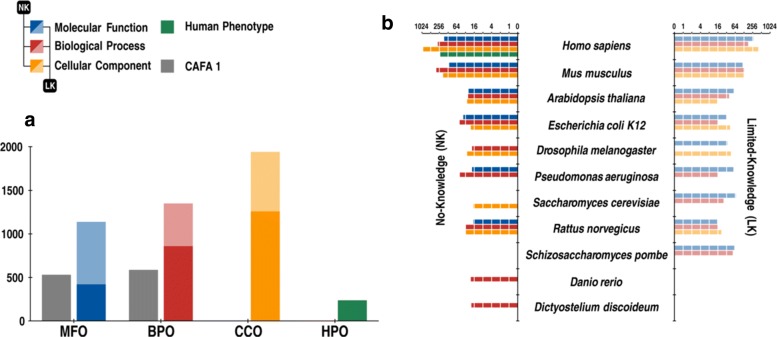
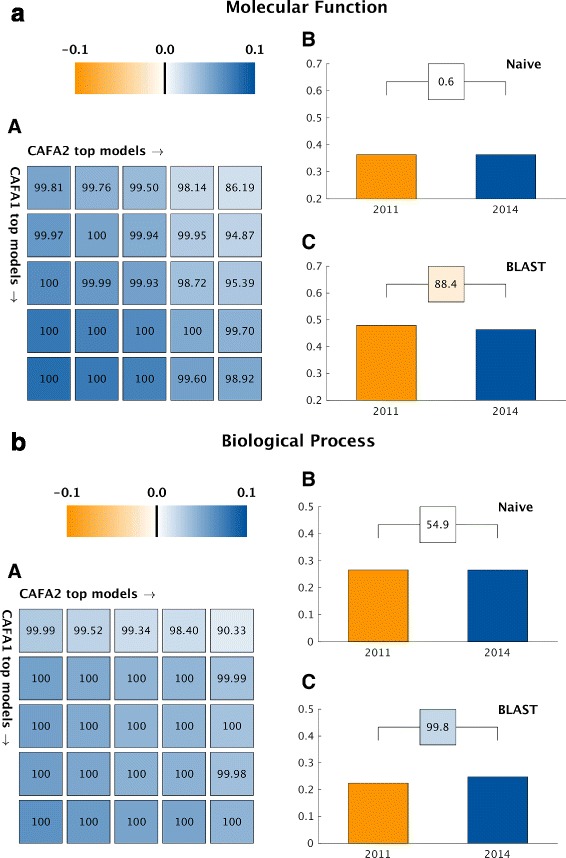
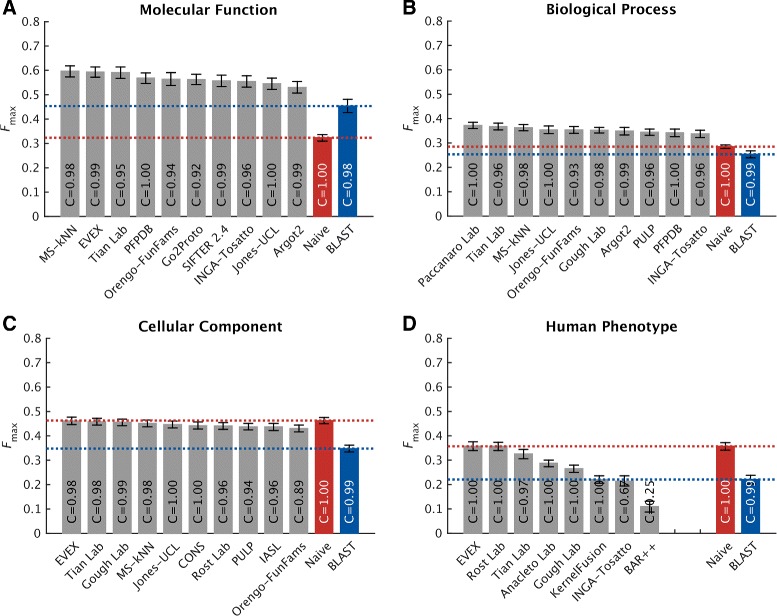
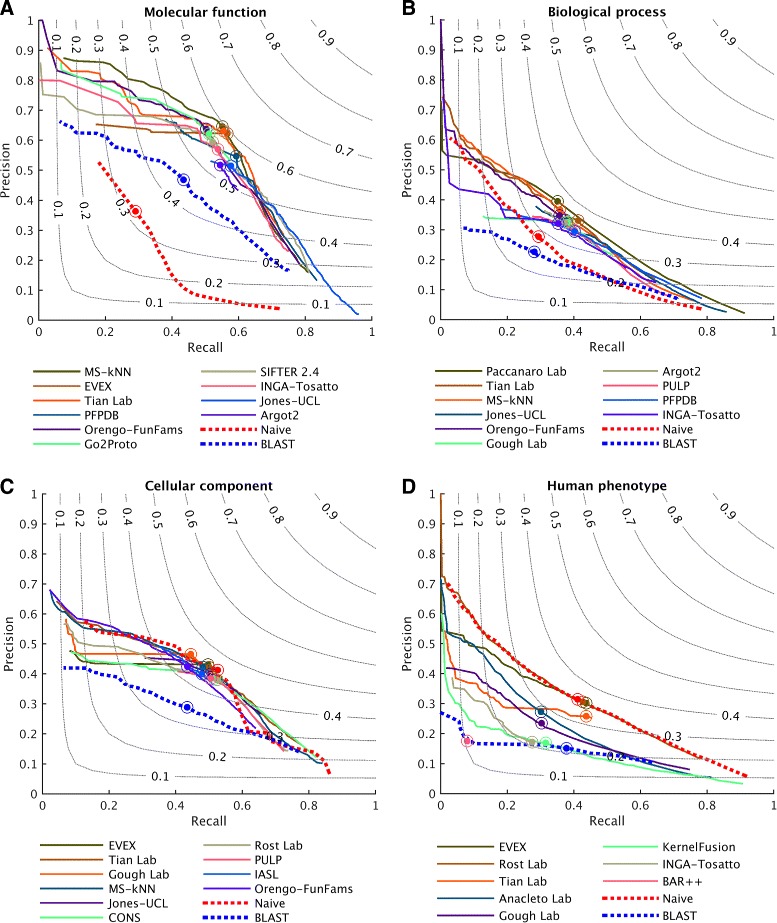
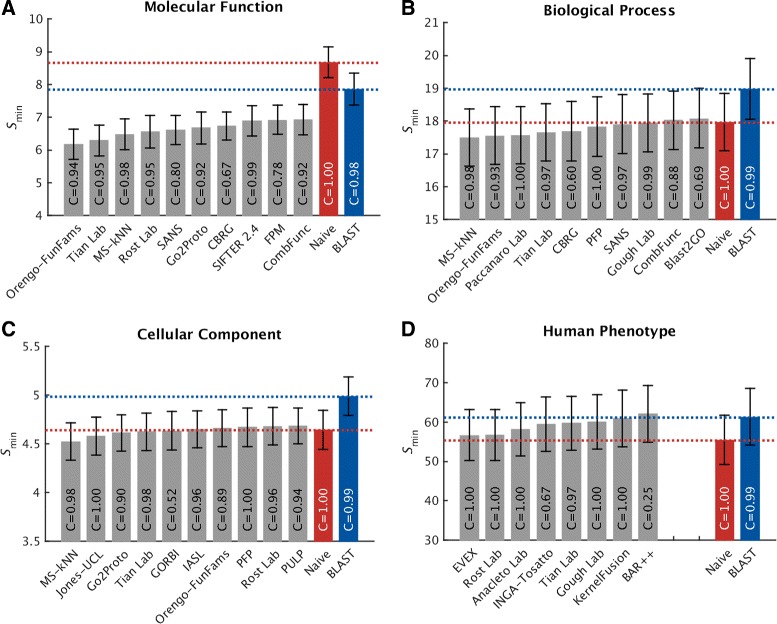
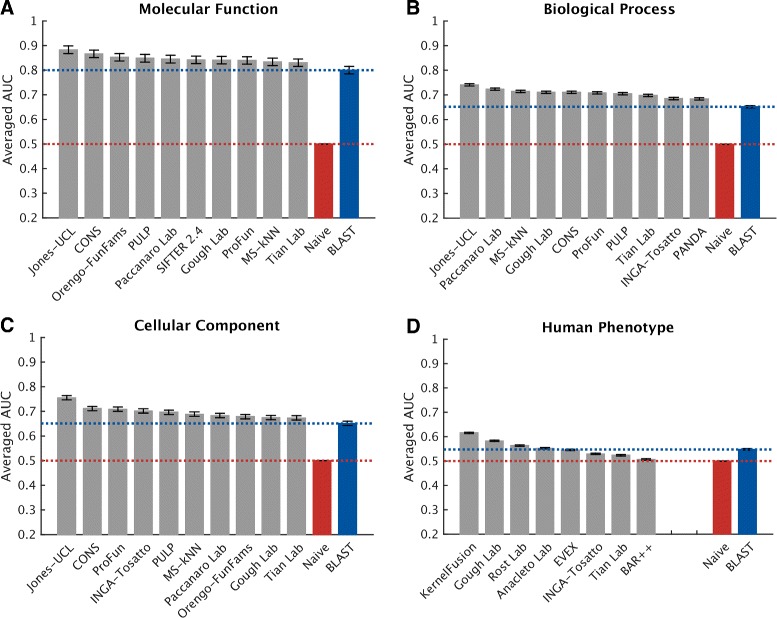
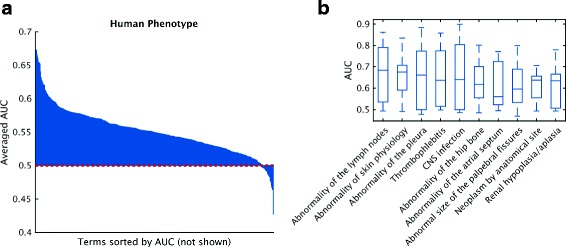
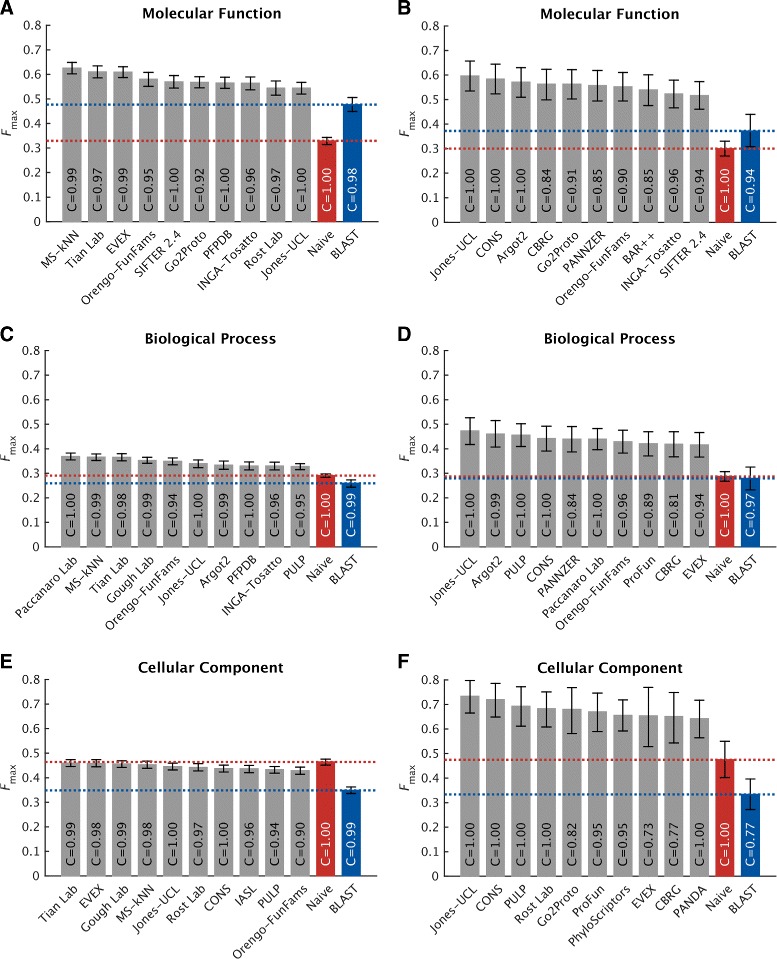
Results: We conducted the second critical assessment of functional annotation (CAFA), a timed challenge to assess computational methods that automatically assign protein function. We evaluated 126 methods from 56 research groups for their ability to predict biological functions using Gene Ontology and gene-disease associations using Human Phenotype Ontology on a set of 3681 proteins from 18 species. CAFA2 featured expanded analysis compared with CAFA1, with regards to data set size, variety, and assessment metrics. To review progress in the field, the analysis compared the best methods from CAFA1 to those of CAFA2.
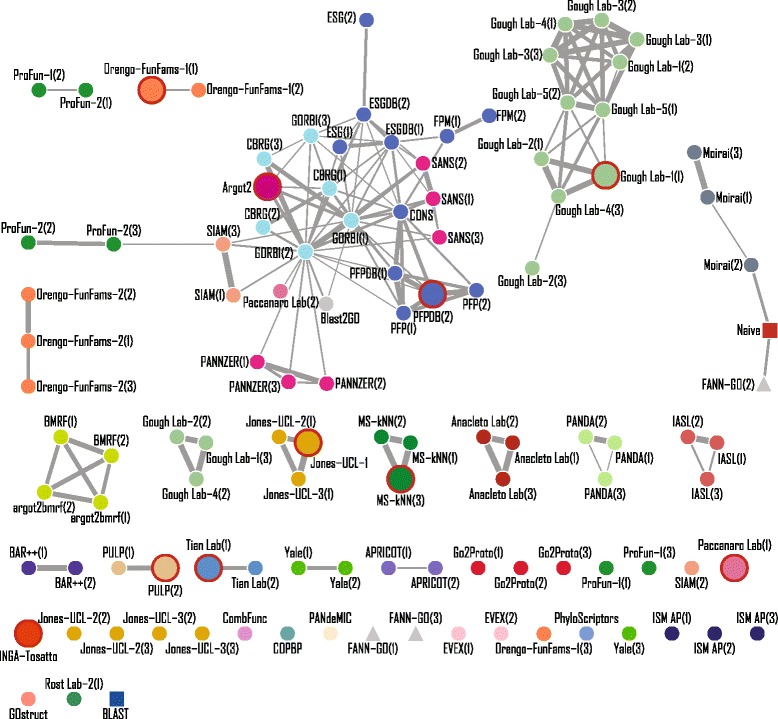
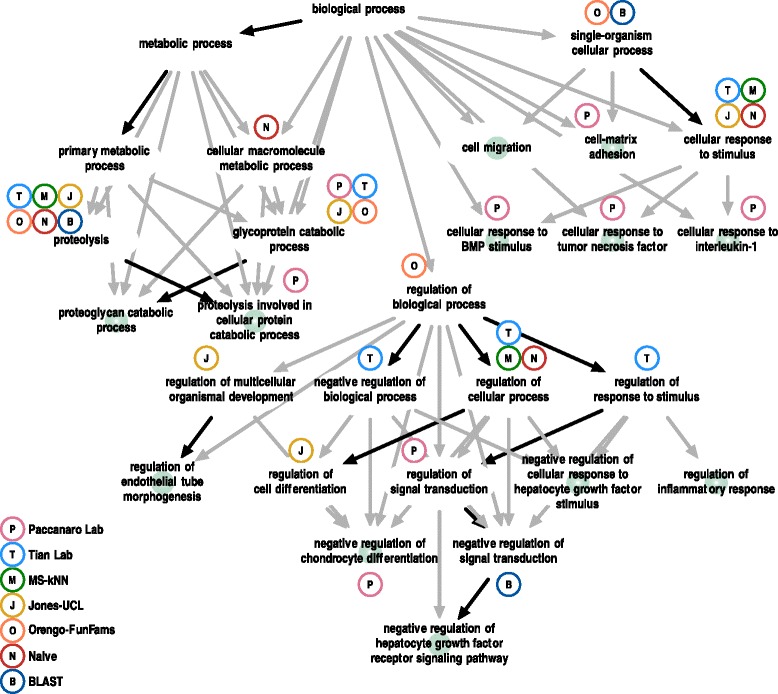
Conclusions: The top-performing methods in CAFA2 outperformed those from CAFA1. This increased accuracy can be attributed to a combination of the growing number of experimental annotations and improved methods for function prediction. The assessment also revealed that the definition of top-performing algorithms is ontology specific, that different performance metrics can be used to probe the nature of accurate predictions, and the relative diversity of predictions in the biological process and human phenotype ontologies. While there was methodological improvement between CAFA1 and CAFA2, the interpretation of results and usefulness of individual methods remain context-dependent.
Keywords: Disease gene prioritization; Protein function prediction.
Figures
References
Publication types
MeSH terms
Substances
Associated data
Grants and funding
- BB/G022771/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/L018241/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- G-1307/PUK_/Parkinson's UK/United Kingdom
- BB/F00964X/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/G022569/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- R01 GM097528/GM/NIGMS NIH HHS/United States
- R01 MH105524/MH/NIMH NIH HHS/United States
- BB/K004131/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- R01 MH111099/MH/NIMH NIH HHS/United States
- BB/L020505/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- R01 GM093123/GM/NIGMS NIH HHS/United States
- R01 GM071749/GM/NIGMS NIH HHS/United States
- BB/F020481/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- R01 LM009722/LM/NLM NIH HHS/United States
- R01 GM076990/GM/NIGMS NIH HHS/United States
- RG/13/5/30112/BHF_/British Heart Foundation/United Kingdom
- R01 GM060595/GM/NIGMS NIH HHS/United States
- BB/M015009/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- UL1 TR002319/TR/NCATS NIH HHS/United States
- BB/L002817/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- UL1 TR000423/TR/NCATS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
