

Heme Oxygenases in Cardiovascular Health and Disease
- PMID: 27604527
- PMCID: PMC5504454
- DOI: 10.1152/physrev.00003.2016
Heme Oxygenases in Cardiovascular Health and Disease
Abstract
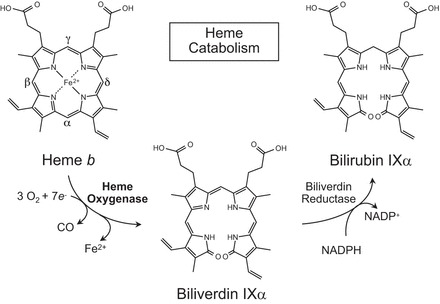
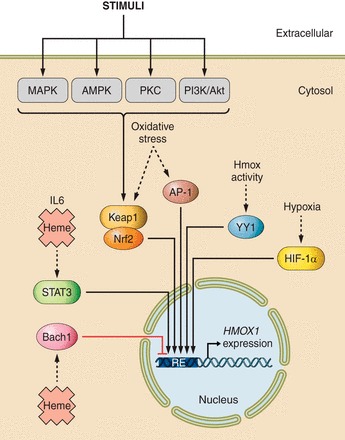
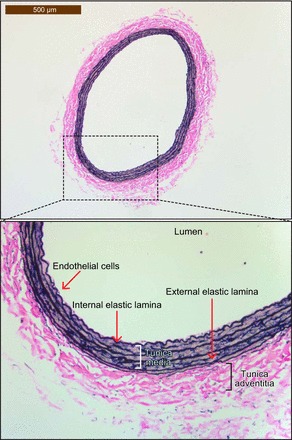
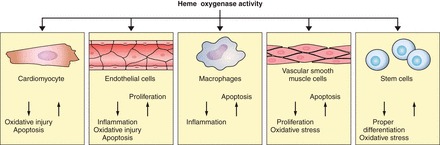
Heme oxygenases are composed of two isozymes, Hmox1 and Hmox2, that catalyze the degradation of heme to carbon monoxide (CO), ferrous iron, and biliverdin, the latter of which is subsequently converted to bilirubin. While initially considered to be waste products, CO and biliverdin/bilirubin have been shown over the last 20 years to modulate key cellular processes, such as inflammation, cell proliferation, and apoptosis, as well as antioxidant defense. This shift in paradigm has led to the importance of heme oxygenases and their products in cell physiology now being well accepted. The identification of the two human cases thus far of heme oxygenase deficiency and the generation of mice deficient in Hmox1 or Hmox2 have reiterated a role for these enzymes in both normal cell function and disease pathogenesis, especially in the context of cardiovascular disease. This review covers the current knowledge on the function of both Hmox1 and Hmox2 at both a cellular and tissue level in the cardiovascular system. Initially, the roles of heme oxygenases in vascular health and the regulation of processes central to vascular diseases are outlined, followed by an evaluation of the role(s) of Hmox1 and Hmox2 in various diseases such as atherosclerosis, intimal hyperplasia, myocardial infarction, and angiogenesis. Finally, the therapeutic potential of heme oxygenases and their products are examined in a cardiovascular disease context, with a focus on how the knowledge we have gained on these enzymes may be capitalized in future clinical studies.
Copyright © 2016 the American Physiological Society.
Figures

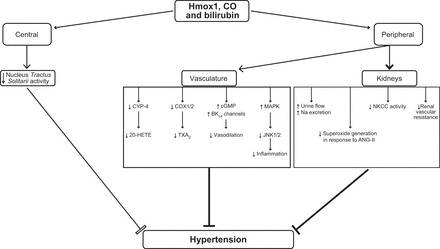
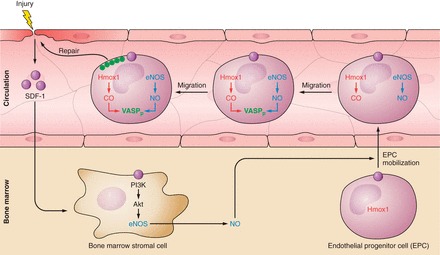
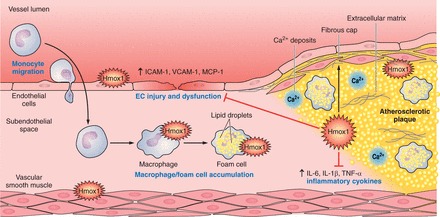
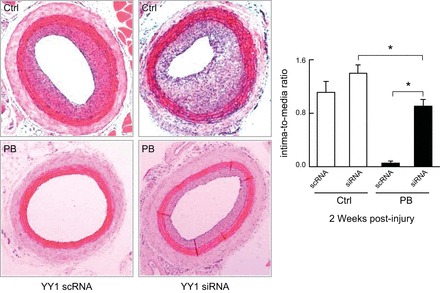
References
-
- Abid S, Houssaini A, Mouraret N, Marcos E, Amsellem V, Wan F, Dubois-Rande JL, Derumeaux G, Boczkowski J, Motterlini R, Adnot S. P21-dependent protective effects of a carbon monoxide-releasing molecule-3 in pulmonary hypertension. Arterioscler Thromb Vasc Biol 34: 304–312, 2014. - PubMed
-
- Abraham NG, Lavrovsky Y, Schwartzman ML, Stoltz RA, Levere RD, Gerritsen ME, Shibahara S, Kappas A. Transfection of the human heme oxygenase gene into rabbit coronary microvessel endothelial cells: protective effect against heme and hemoglobin toxicity. Proc Natl Acad Sci USA 92: 6798–6802, 1995. - PMC - PubMed
-
- Abraham NG, Scapagnini G, Kappas A. Human heme oxygenase: cell cycle-dependent expression and DNA microarray identification of multiple gene responses after transduction of endothelial cells. J Cell Biochem 90: 1098–1111, 2003. - PubMed
-
- Abran D, Dumont I, Hardy P, Peri K, Li DY, Molotchnikoff S, Varma DR, Chemtob S. Characterization and regulation of prostaglandin E2 receptor and receptor-coupled functions in the choroidal vasculature of the pig during development. Circ Res 80: 463–472, 1997. - PubMed
-
- Agarwal A, Balla J, Balla G, Croatt AJ, Vercellotti GM, Nath KA. Renal tubular epithelial cells mimic endothelial cells upon exposure to oxidized LDL. Am J Physiol Renal Fluid Electrolyte Physiol 271: F814–F823, 1996. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources