

Predicting the Stability of Homologous Gene Duplications in a Plant RNA Virus
- PMID: 27604880
- PMCID: PMC5633665
- DOI: 10.1093/gbe/evw219
Predicting the Stability of Homologous Gene Duplications in a Plant RNA Virus
Abstract
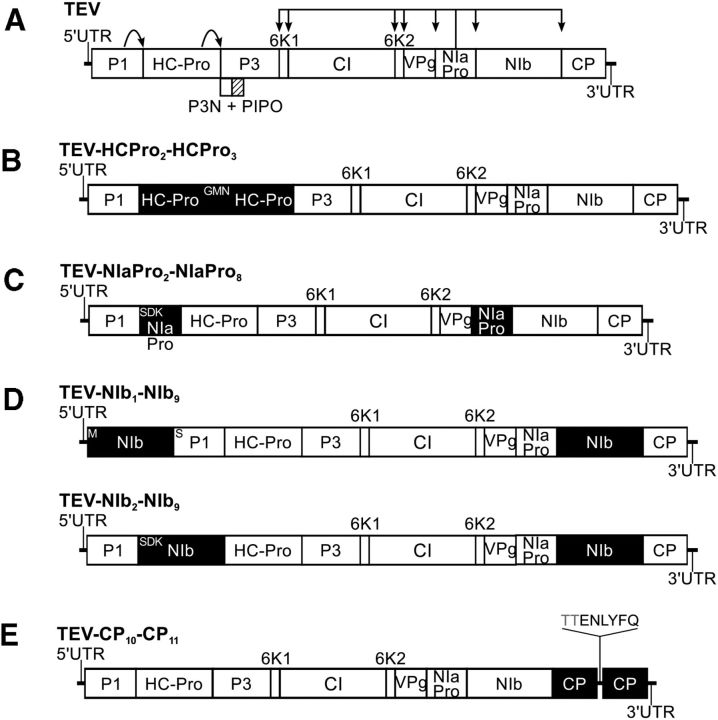
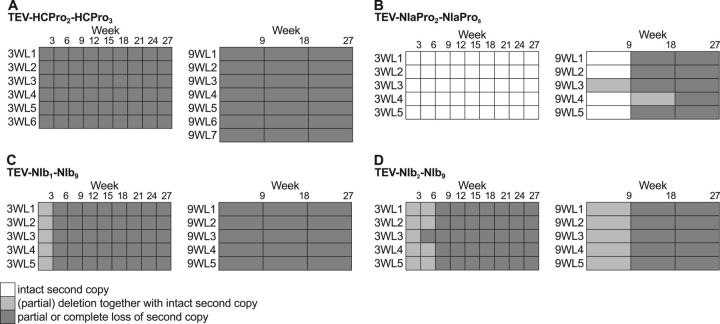
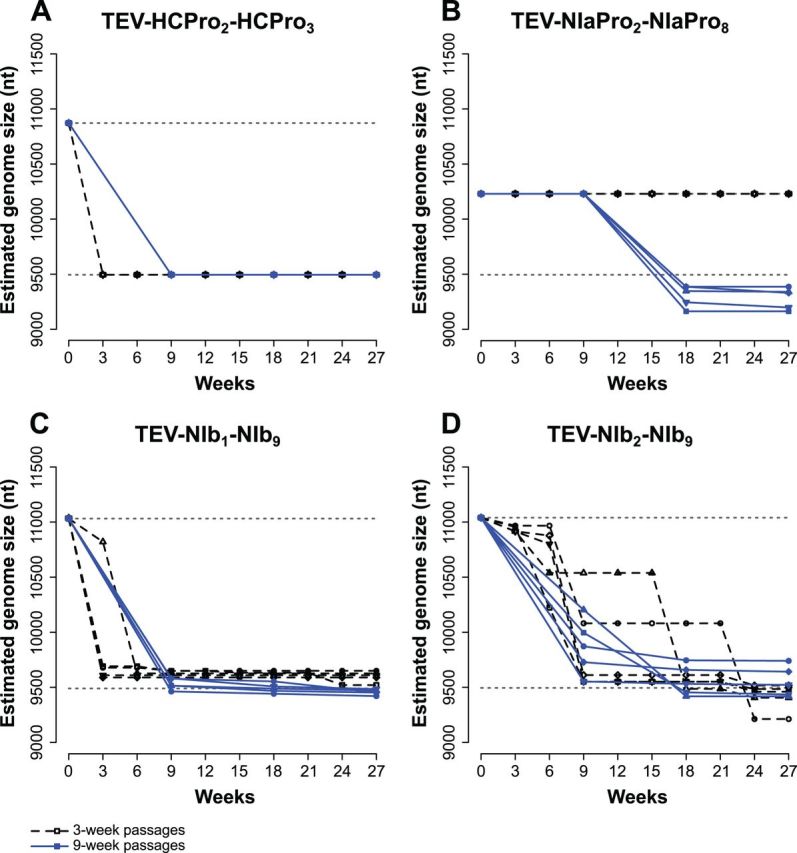
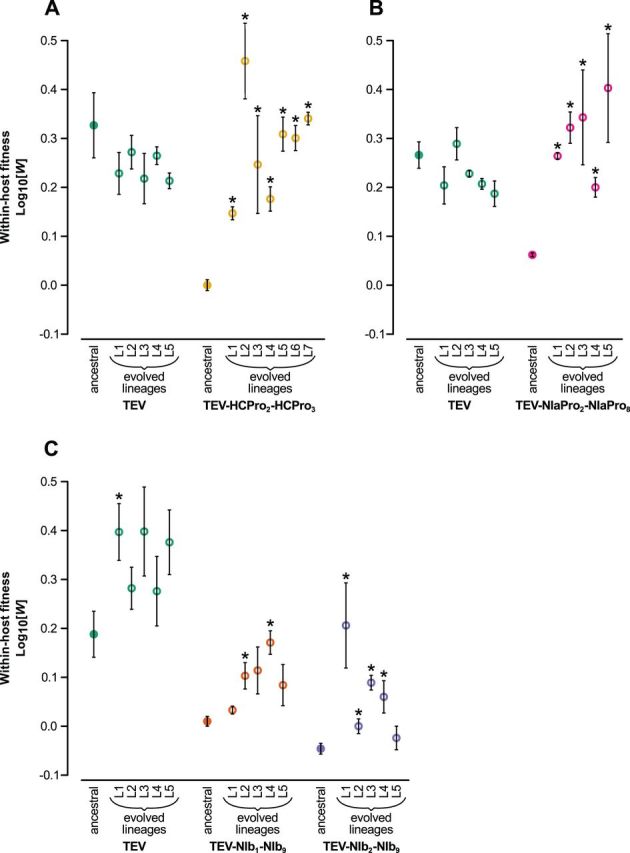
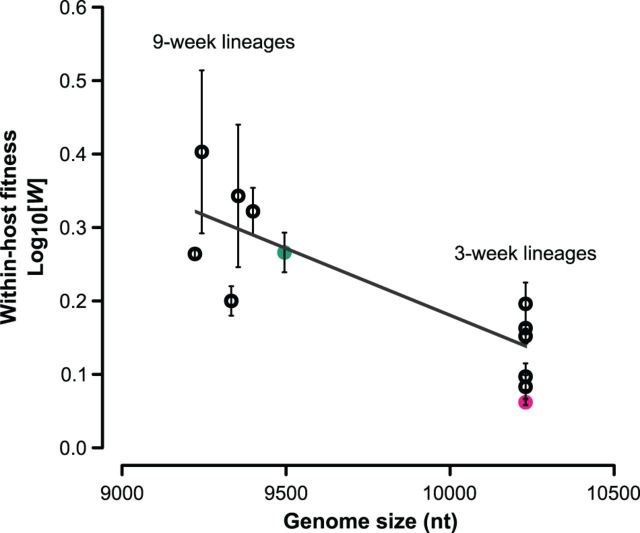
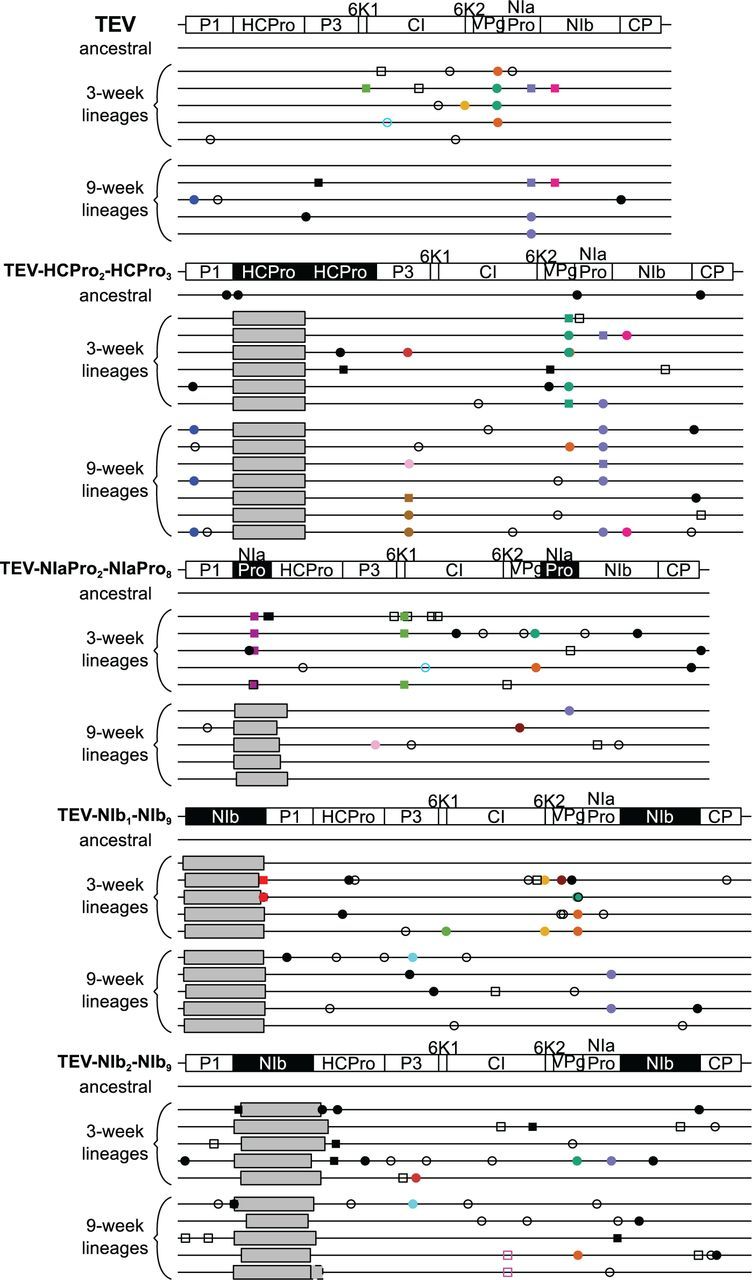
One of the striking features of many eukaryotes is the apparent amount of redundancy in coding and non-coding elements of their genomes. Despite the possible evolutionary advantages, there are fewer examples of redundant sequences in viral genomes, particularly those with RNA genomes. The factors constraining the maintenance of redundant sequences in present-day RNA virus genomes are not well known. Here, we use Tobacco etch virus, a plant RNA virus, to investigate the stability of genetically redundant sequences by generating viruses with potentially beneficial gene duplications. Subsequently, we tested the viability of these viruses and performed experimental evolution. We found that all gene duplication events resulted in a loss of viability or in a significant reduction in viral fitness. Moreover, upon analyzing the genomes of the evolved viruses, we always observed the deletion of the duplicated gene copy and maintenance of the ancestral copy. Interestingly, there were clear differences in the deletion dynamics of the duplicated gene associated with the passage duration and the size and position of the duplicated copy. Based on the experimental data, we developed a mathematical model to characterize the stability of genetically redundant sequences, and showed that fitness effects are not enough to predict genomic stability. A context-dependent recombination rate is also required, with the context being the duplicated gene and its position. Our results therefore demonstrate experimentally the deleterious nature of gene duplications in RNA viruses. Beside previously described constraints on genome size, we identified additional factors that reduce the likelihood of the maintenance of duplicated genes.
Keywords: experimental evolution; gene duplication; genome stability; virus evolution.
© The Author 2016. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures

Similar articles
-
Multiple Barriers to the Evolution of Alternative Gene Orders in a Positive-Strand RNA Virus.Genetics. 2016 Apr;202(4):1503-21. doi: 10.1534/genetics.115.185017. Epub 2016 Feb 11. Genetics. 2016. PMID: 26868766 Free PMC article.
-
Gene duplication is infrequent in the recent evolutionary history of RNA viruses.Mol Biol Evol. 2013 Jun;30(6):1263-9. doi: 10.1093/molbev/mst044. Epub 2013 Mar 13. Mol Biol Evol. 2013. PMID: 23486612
-
Experimental evolution of pseudogenization and gene loss in a plant RNA virus.Mol Biol Evol. 2014 Jan;31(1):121-34. doi: 10.1093/molbev/mst175. Epub 2013 Oct 8. Mol Biol Evol. 2014. PMID: 24109604 Free PMC article.
-
How RNA viruses maintain their genome integrity.J Gen Virol. 2010 Jun;91(Pt 6):1373-87. doi: 10.1099/vir.0.020818-0. Epub 2010 Mar 24. J Gen Virol. 2010. PMID: 20335491 Review.
-
Cis-acting RNA elements in positive-strand RNA plant virus genomes.Virology. 2015 May;479-480:434-43. doi: 10.1016/j.virol.2015.02.032. Epub 2015 Mar 7. Virology. 2015. PMID: 25759098 Review.
Cited by
-
P1 of Sweet Potato Feathery Mottle Virus Shows Strong Adaptation Capacity, Replacing P1-HCPro in a Chimeric Plum Pox Virus.J Virol. 2021 Jun 24;95(14):e0015021. doi: 10.1128/JVI.00150-21. Epub 2021 Jun 24. J Virol. 2021. PMID: 33952634 Free PMC article.
-
Population dynamics of synthetic terraformation motifs.R Soc Open Sci. 2018 Jul 4;5(7):180121. doi: 10.1098/rsos.180121. eCollection 2018 Jul. R Soc Open Sci. 2018. PMID: 30109068 Free PMC article.
-
Emergency Services of Viral RNAs: Repair and Remodeling.Microbiol Mol Biol Rev. 2018 Mar 14;82(2):e00067-17. doi: 10.1128/MMBR.00067-17. Print 2018 Jun. Microbiol Mol Biol Rev. 2018. PMID: 29540453 Free PMC article. Review.
-
Viral Fitness Correlates with the Magnitude and Direction of the Perturbation Induced in the Host's Transcriptome: The Tobacco Etch Potyvirus-Tobacco Case Study.Mol Biol Evol. 2018 Jul 1;35(7):1599-1615. doi: 10.1093/molbev/msy038. Mol Biol Evol. 2018. PMID: 29562354 Free PMC article.
-
High virulence does not necessarily impede viral adaptation to a new host: a case study using a plant RNA virus.BMC Evol Biol. 2017 Jan 19;17(1):25. doi: 10.1186/s12862-017-0881-7. BMC Evol Biol. 2017. PMID: 28103791 Free PMC article.
References
-
- Andersson DI, Hughes D. 2009. Gene amplification and adaptive evolution in bacteria. Annu. Rev. Genet. 43:167–195. - PubMed
-
- Atreya CD, Atreya PL, Thornbury DW, Pirone TP. 1992. Site-directed mutations in the potyvirus HC-PRO gene affect helper component activity, virus accumulation, and symptom expression in infected tobacco plants. Virology 191:106–111. - PubMed
-
- Bedoya LC, Daròs J-A. 2010. Stability of Tobacco etch virus infectious clones in plasmid vectors. Virus Res. 149:234–240. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
