

Rare variant phasing and haplotypic expression from RNA sequencing with phASER
- PMID: 27605262
- PMCID: PMC5025529
- DOI: 10.1038/ncomms12817
Rare variant phasing and haplotypic expression from RNA sequencing with phASER
Abstract
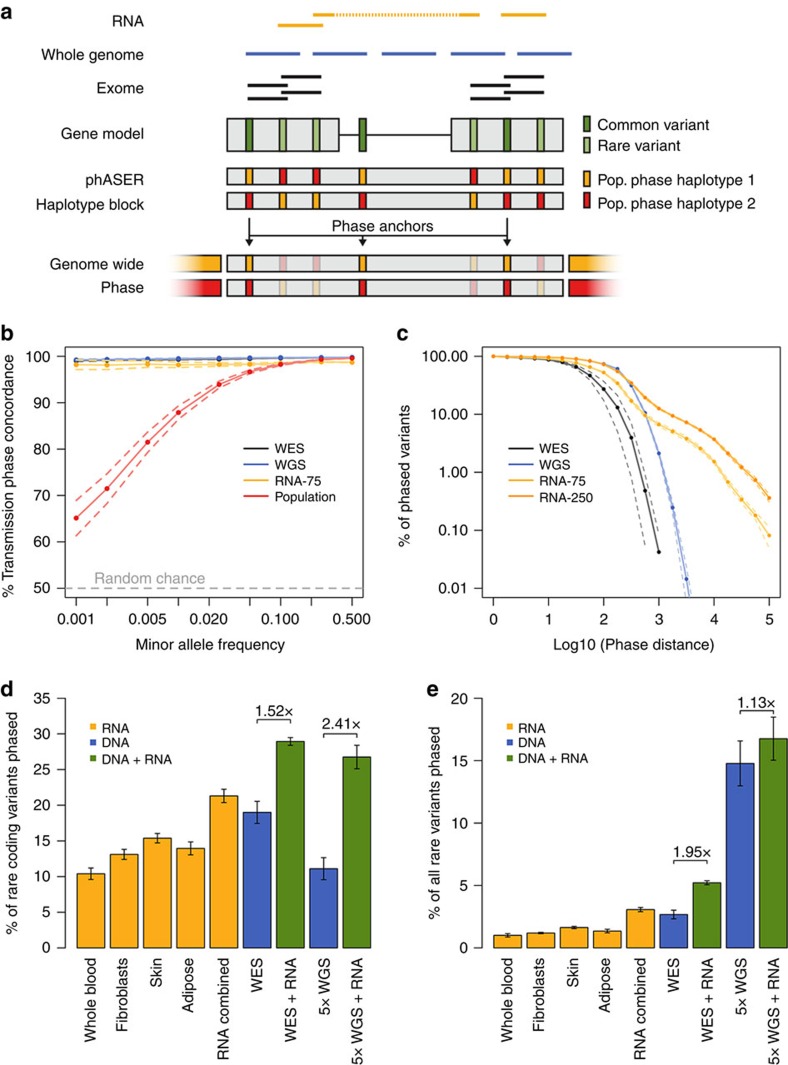
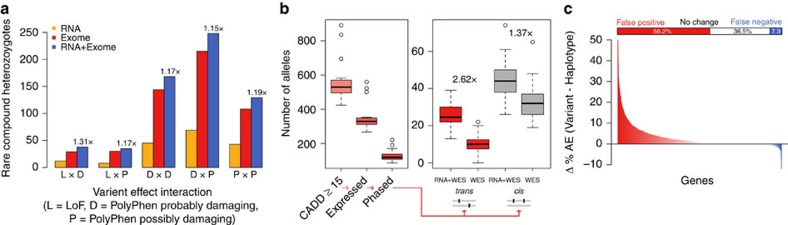
Haplotype phasing of genetic variants is important for clinical interpretation of the genome, population genetic analysis and functional genomic analysis of allelic activity. Here we present phASER, an accurate approach for phasing variants that are overlapped by sequencing reads, including those from RNA sequencing (RNA-seq), which often span multiple exons due to splicing. Using diverse RNA-seq data we demonstrate that this provides more accurate phasing of rare variants compared with population-based phasing and allows phasing of variants in the same gene up to hundreds of kilobases away that cannot be obtained from DNA sequencing (DNA-seq) reads. We show that in the context of medical genetic studies this improves the resolution of compound heterozygotes. Additionally, phASER provides measures of haplotypic expression that increase power and accuracy in studies of allelic expression. In summary, phasing using RNA-seq and phASER is accurate and improves studies where rare variant haplotypes or allelic expression is needed.
Figures
References
-
- Delaneau O., Marchini J. & Zagury J.-F. A linear complexity phasing method for thousands of genomes. Nat. Methods 9, 179–181 (2012). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 DA006227/DA/NIDA NIH HHS/United States
- R01 HD057036/HD/NICHD NIH HHS/United States
- R01 MH101782/MH/NIMH NIH HHS/United States
- R01 MH101810/MH/NIMH NIH HHS/United States
- R01 MH101819/MH/NIMH NIH HHS/United States
- R01 MH090936/MH/NIMH NIH HHS/United States
- R01 MH090951/MH/NIMH NIH HHS/United States
- R01 MH101820/MH/NIMH NIH HHS/United States
- P30 DK026687/DK/NIDDK NIH HHS/United States
- R01 MH101822/MH/NIMH NIH HHS/United States
- UL1 RR024156/RR/NCRR NIH HHS/United States
- R01 DA033684/DA/NIDA NIH HHS/United States
- R01 MH106842/MH/NIMH NIH HHS/United States
- R01 MH101825/MH/NIMH NIH HHS/United States
- R01 MH090948/MH/NIMH NIH HHS/United States
- R01 MH090941/MH/NIMH NIH HHS/United States
- R01 GM122924/GM/NIGMS NIH HHS/United States
- HHSN261200800001C/RC/CCR NIH HHS/United States
- R01 MH090937/MH/NIMH NIH HHS/United States
- HHSN268201000029C/HL/NHLBI NIH HHS/United States
- HHSN261200800001E/CA/NCI NIH HHS/United States
- R01 MH101814/MH/NIMH NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
