

HIPSTR and thousands of lncRNAs are heterogeneously expressed in human embryos, primordial germ cells and stable cell lines
- PMID: 27605307
- PMCID: PMC5015059
- DOI: 10.1038/srep32753
HIPSTR and thousands of lncRNAs are heterogeneously expressed in human embryos, primordial germ cells and stable cell lines
Erratum in
-
Corrigendum: HIPSTR and thousands of lncRNAs are heterogeneously expressed in human embryos, primordial germ cells and stable cell lines.Sci Rep. 2017 Mar 16;7:44632. doi: 10.1038/srep44632. Sci Rep. 2017. PMID: 28300214 Free PMC article. No abstract available.
Abstract
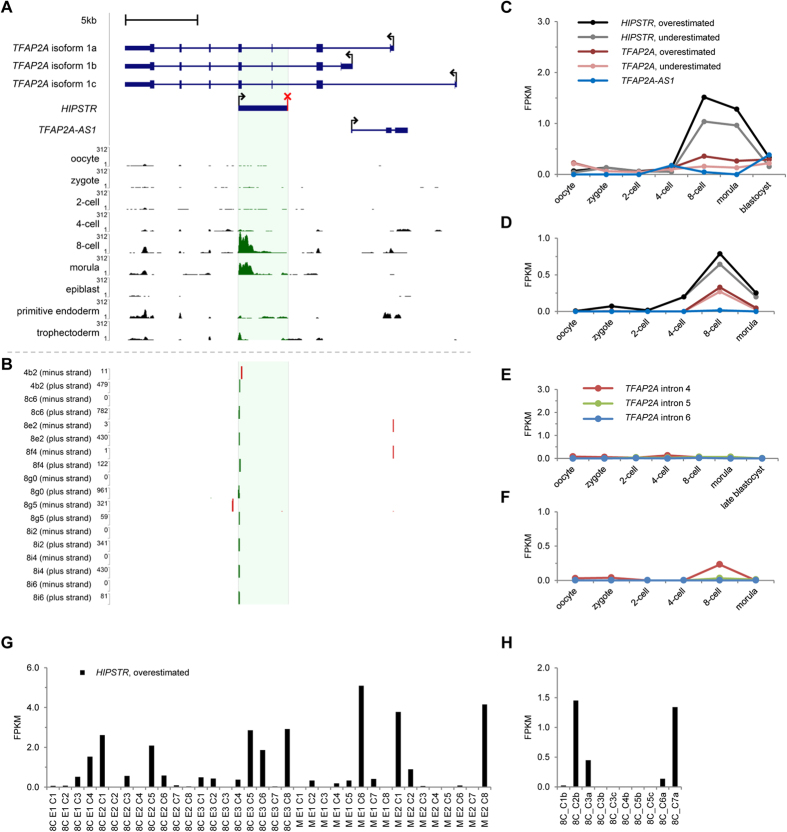
Eukaryotic genomes are transcribed into numerous regulatory long non-coding RNAs (lncRNAs). Compared to mRNAs, lncRNAs display higher developmental stage-, tissue-, and cell-subtype-specificity of expression, and are generally less abundant in a population of cells. Despite the progress in single-cell-focused research, the origins of low population-level expression of lncRNAs in homogeneous populations of cells are poorly understood. Here, we identify HIPSTR (Heterogeneously expressed from the Intronic Plus Strand of the TFAP2A-locus RNA), a novel lncRNA gene in the developmentally regulated TFAP2A locus. HIPSTR has evolutionarily conserved expression patterns, its promoter is most active in undifferentiated cells, and depletion of HIPSTR in HEK293 and in pluripotent H1BP cells predominantly affects the genes involved in early organismal development and cell differentiation. Most importantly, we find that HIPSTR is specifically induced and heterogeneously expressed in the 8-cell-stage human embryos during the major wave of embryonic genome activation. We systematically explore the phenomenon of cell-to-cell variation of gene expression and link it to low population-level expression of lncRNAs, showing that, similar to HIPSTR, the expression of thousands of lncRNAs is more highly heterogeneous than the expression of mRNAs in the individual, otherwise indistinguishable cells of totipotent human embryos, primordial germ cells, and stable cell lines.
Figures




References
-
- Quinn J. J. & Chang H. Y. Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet 17, 47–62 (2015). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
