

Glycolysis selectively shapes the presynaptic action potential waveform
- PMID: 27605535
- PMCID: PMC5133309
- DOI: 10.1152/jn.00629.2016
Glycolysis selectively shapes the presynaptic action potential waveform
Abstract
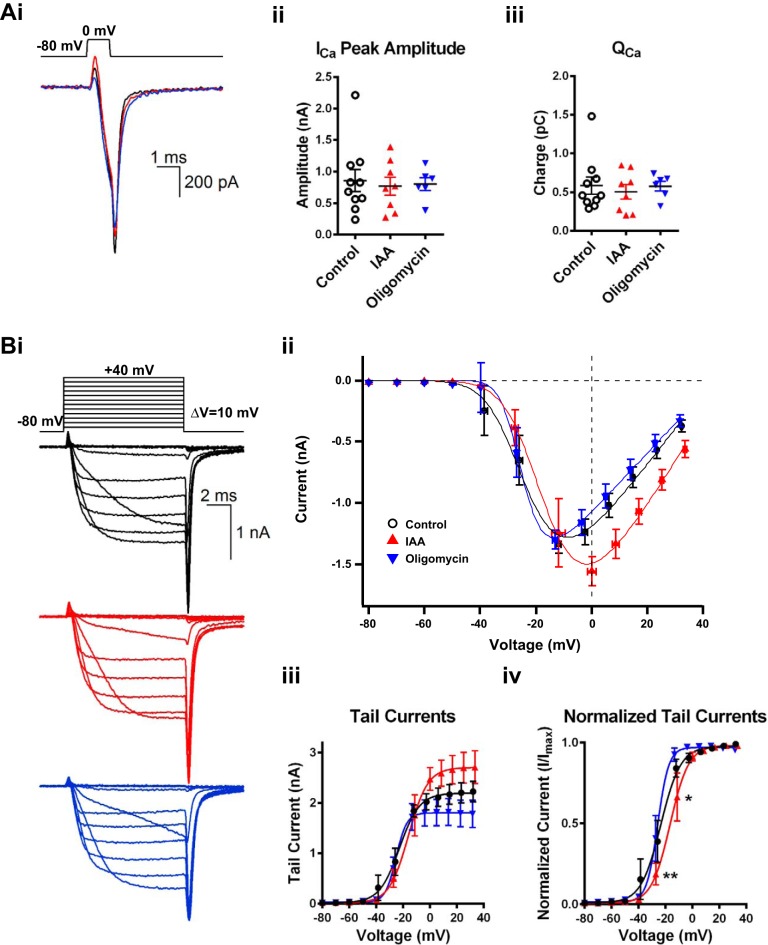
Mitochondria are major suppliers of cellular energy in neurons; however, utilization of energy from glycolysis vs. mitochondrial oxidative phosphorylation (OxPhos) in the presynaptic compartment during neurotransmission is largely unknown. Using presynaptic and postsynaptic recordings from the mouse calyx of Held, we examined the effect of acute selective pharmacological inhibition of glycolysis or mitochondrial OxPhos on multiple mechanisms regulating presynaptic function. Inhibition of glycolysis via glucose depletion and iodoacetic acid (1 mM) treatment, but not mitochondrial OxPhos, rapidly altered transmission, resulting in highly variable, oscillating responses. At reduced temperature, this same treatment attenuated synaptic transmission because of a smaller and broader presynaptic action potential (AP) waveform. We show via experimental manipulation and ion channel modeling that the altered AP waveform results in smaller Ca2+ influx, resulting in attenuated excitatory postsynaptic currents (EPSCs). In contrast, inhibition of mitochondria-derived ATP production via extracellular pyruvate depletion and bath-applied oligomycin (1 μM) had no significant effect on Ca2+ influx and did not alter the AP waveform within the same time frame (up to 30 min), and the resultant EPSC remained unaffected. Glycolysis, but not mitochondrial OxPhos, is thus required to maintain basal synaptic transmission at the presynaptic terminal. We propose that glycolytic enzymes are closely apposed to ATP-dependent ion pumps on the presynaptic membrane. Our results indicate a novel mechanism for the effect of hypoglycemia on neurotransmission. Attenuated transmission likely results from a single presynaptic mechanism at reduced temperature: a slower, smaller AP, before and independent of any effect on synaptic vesicle release or receptor activity.
Keywords: bioenergetics; calyx of Held; hypoglycemia; oxidative phosphorylation; stroke.
Copyright © 2016 the American Physiological Society.
Figures










References
-
- Alle H, Roth A, Geiger JR. Energy-efficient action potentials in hippocampal mossy fibers. Science 325: 1405–1408, 2009. - PubMed
-
- Ames A 3rd, Gurian BS. Effects of glucose and oxygen deprivation on function of isolated mammalian retina. J Neurophysiol 26: 617–634, 1963. - PubMed
-
- Awatramani GB, Price GD, Trussell LO. Modulation of transmitter release by presynaptic resting potential and background calcium levels. Neuron 48: 109–121, 2005. - PubMed
-
- Bak LK, Schousboe A, Sonnewald U, Waagepetersen HS. Glucose is necessary to maintain neurotransmitter homeostasis during synaptic activity in cultured glutamatergic neurons. J Cereb Blood Flow Metab 26: 1285–1297, 2006. - PubMed
-
- Bak LK, Walls AB, Schousboe A, Ring A, Sonnewald U, Waagepetersen HS. Neuronal glucose but not lactate utilization is positively correlated with NMDA-induced neurotransmission and fluctuations in cytosolic Ca2+ levels. J Neurochem 109: 87–93, 2009. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous