

Two locus inheritance of non-syndromic midline craniosynostosis via rare SMAD6 and common BMP2 alleles
- PMID: 27606499
- PMCID: PMC5045293
- DOI: 10.7554/eLife.20125
Two locus inheritance of non-syndromic midline craniosynostosis via rare SMAD6 and common BMP2 alleles
Abstract
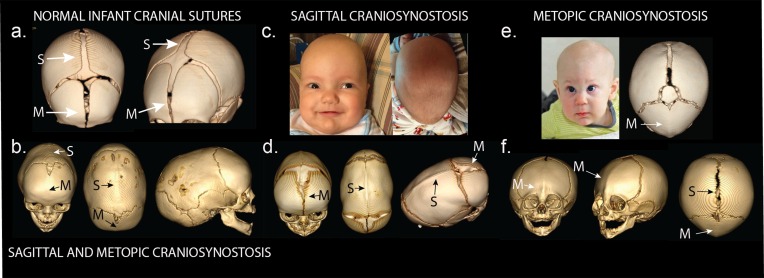
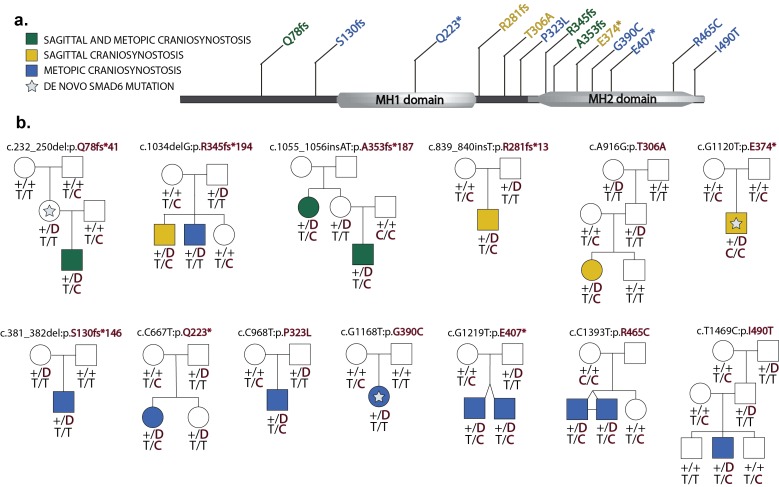
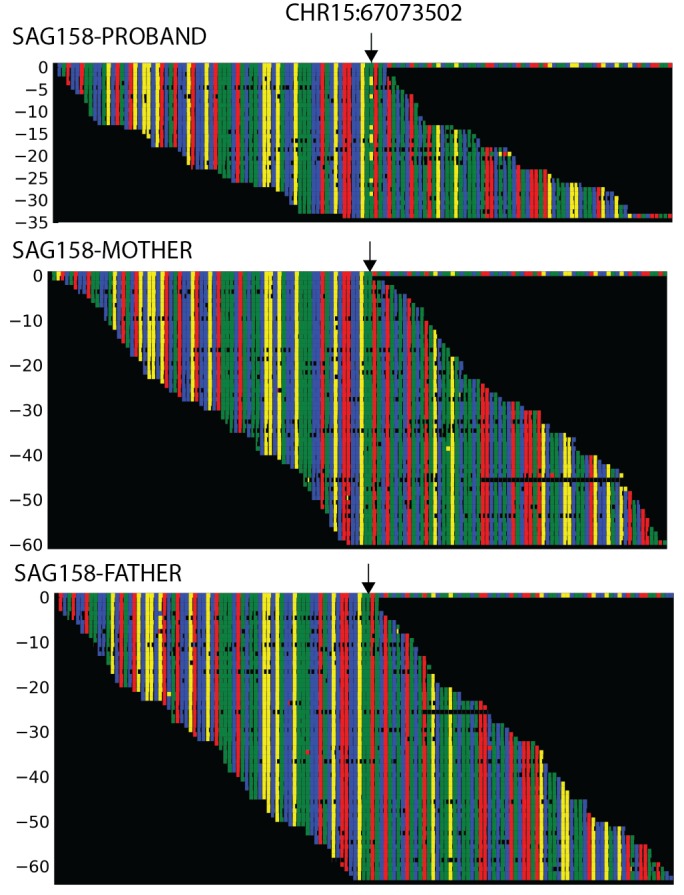
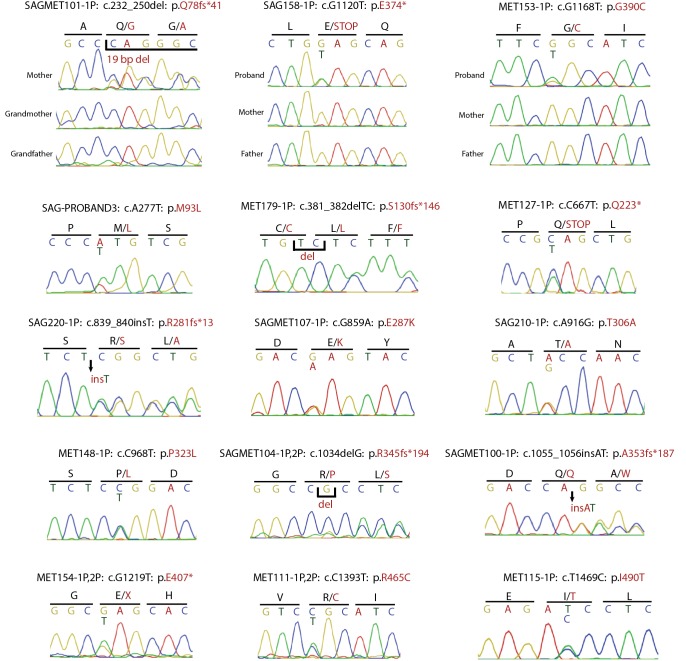
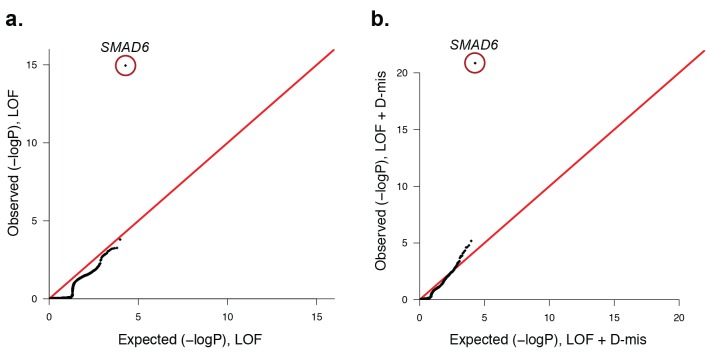
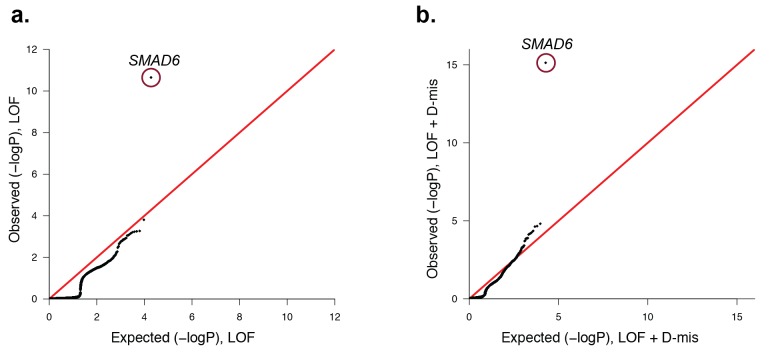
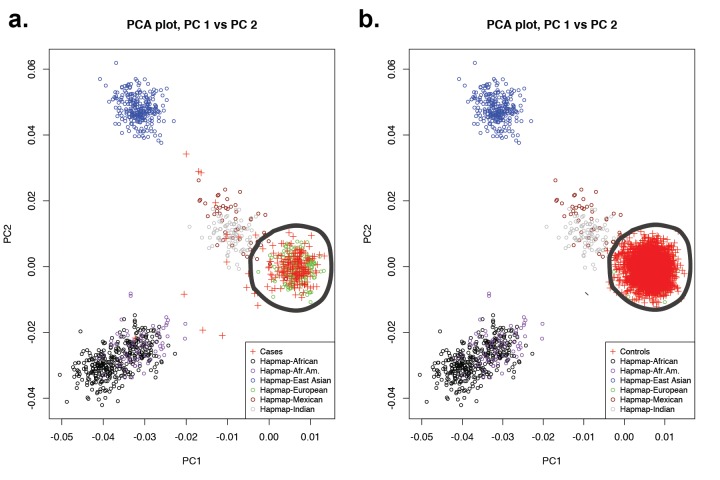
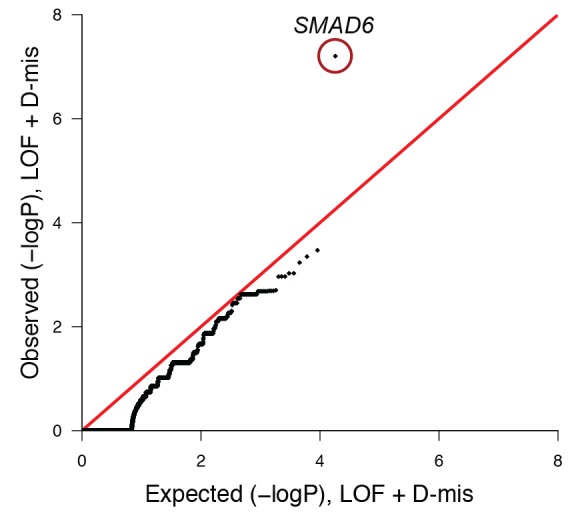
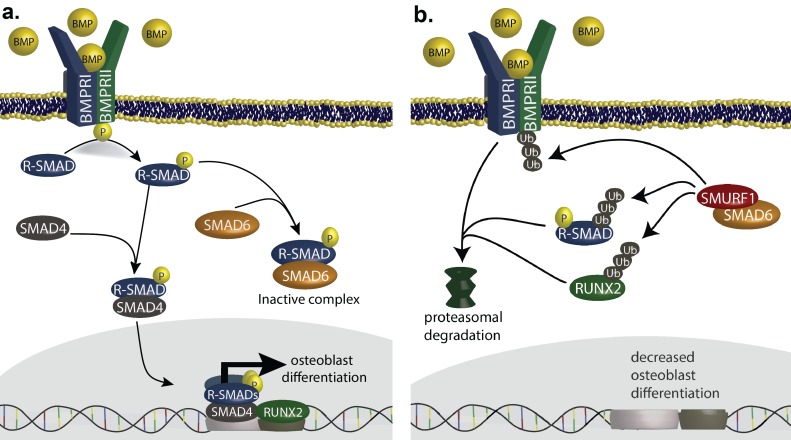
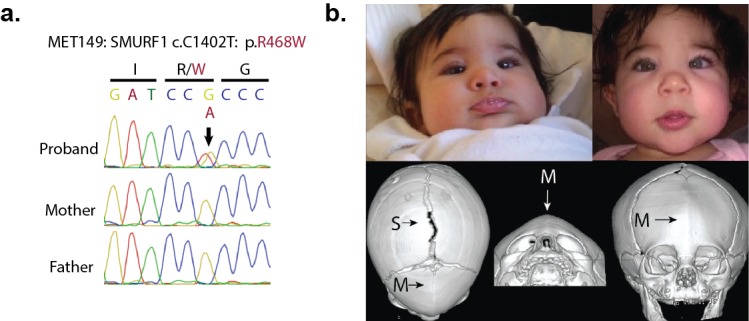
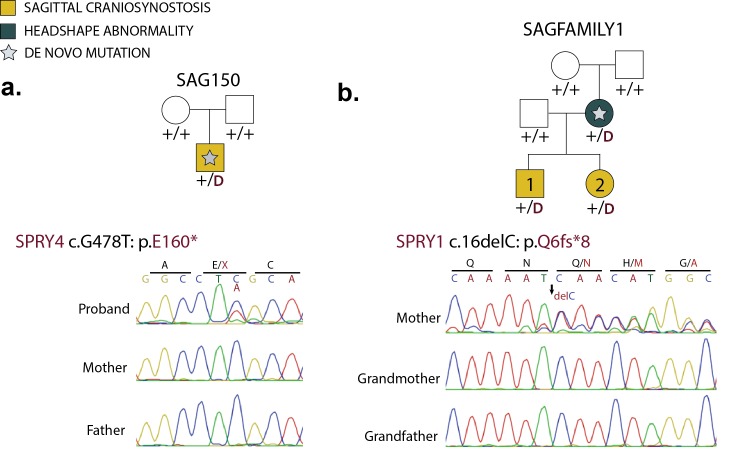
Premature fusion of the cranial sutures (craniosynostosis), affecting 1 in 2000 newborns, is treated surgically in infancy to prevent adverse neurologic outcomes. To identify mutations contributing to common non-syndromic midline (sagittal and metopic) craniosynostosis, we performed exome sequencing of 132 parent-offspring trios and 59 additional probands. Thirteen probands (7%) had damaging de novo or rare transmitted mutations in SMAD6, an inhibitor of BMP - induced osteoblast differentiation (p<10-20). SMAD6 mutations nonetheless showed striking incomplete penetrance (<60%). Genotypes of a common variant near BMP2 that is strongly associated with midline craniosynostosis explained nearly all the phenotypic variation in these kindreds, with highly significant evidence of genetic interaction between these loci via both association and analysis of linkage. This epistatic interaction of rare and common variants defines the most frequent cause of midline craniosynostosis and has implications for the genetic basis of other diseases.
Keywords: BMP2; SMAD6; SMURF1; SPRY1; SPRY4; chromosomes; craniofacial; craniosynostosis; de novo mutation; evolutionary biology; exome sequencing; genes; genomics; human; human genetics; incomplete penetrance.
Conflict of interest statement
The authors declare that no competing interests exist.
Figures
Comment in
- doi: 10.7554/eLife.21162
References
-
- Dathe K, Kjaer KW, Brehm A, Meinecke P, Nürnberg P, Neto JC, Brunoni D, Tommerup N, Ott CE, Klopocki E, Seemann P, Mundlos S. Duplications involving a conserved regulatory element downstream of BMP2 are associated with brachydactyly type A2. American Journal of Human Genetics. 2009;84:483–492. doi: 10.1016/j.ajhg.2009.03.001. - DOI - PMC - PubMed
-
- De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, Kou Y, Liu L, Fromer M, Walker S, Singh T, Klei L, Kosmicki J, Shih-Chen F, Aleksic B, Biscaldi M, Bolton PF, Brownfeld JM, Cai J, Campbell NG, Carracedo A, Chahrour MH, Chiocchetti AG, Coon H, Crawford EL, Curran SR, Dawson G, Duketis E, Fernandez BA, Gallagher L, Geller E, Guter SJ, Hill RS, Ionita-Laza J, Jimenz Gonzalez P, Kilpinen H, Klauck SM, Kolevzon A, Lee I, Lei I, Lei J, Lehtimäki T, Lin CF, Ma'ayan A, Marshall CR, McInnes AL, Neale B, Owen MJ, Ozaki N, Parellada M, Parr JR, Purcell S, Puura K, Rajagopalan D, Rehnström K, Reichenberg A, Sabo A, Sachse M, Sanders SJ, Schafer C, Schulte-Rüther M, Skuse D, Stevens C, Szatmari P, Tammimies K, Valladares O, Voran A, Li-San W, Weiss LA, Willsey AJ, Yu TW, Yuen RK, Cook EH, Freitag CM, Gill M, Hultman CM, Lehner T, Palotie A, Schellenberg GD, Sklar P, State MW, Sutcliffe JS, Walsh CA, Scherer SW, Zwick ME, Barett JC, Cutler DJ, Roeder K, Devlin B, Daly MJ, Buxbaum JD, DDD Study. Homozygosity Mapping Collaborative for Autism. UK10K Consortium Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515:209–215. doi: 10.1038/nature13772. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
