

Domestication and Divergence of Saccharomyces cerevisiae Beer Yeasts
- PMID: 27610566
- PMCID: PMC5018251
- DOI: 10.1016/j.cell.2016.08.020
Domestication and Divergence of Saccharomyces cerevisiae Beer Yeasts
Abstract
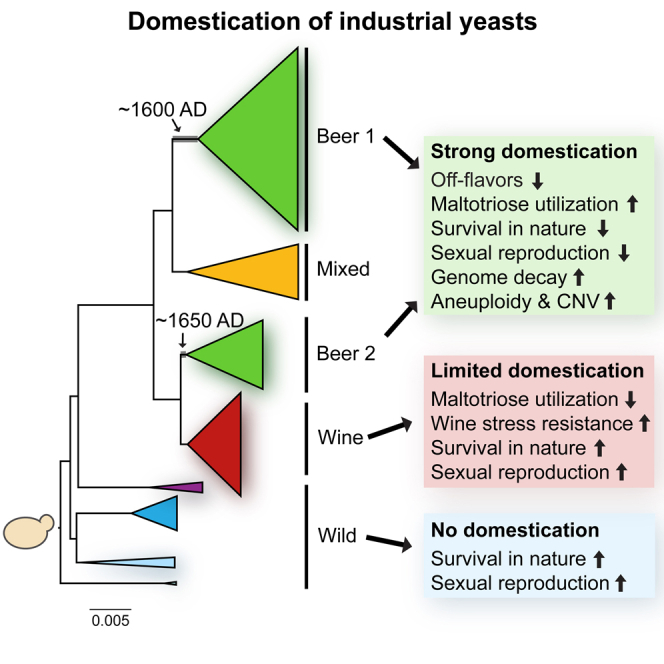
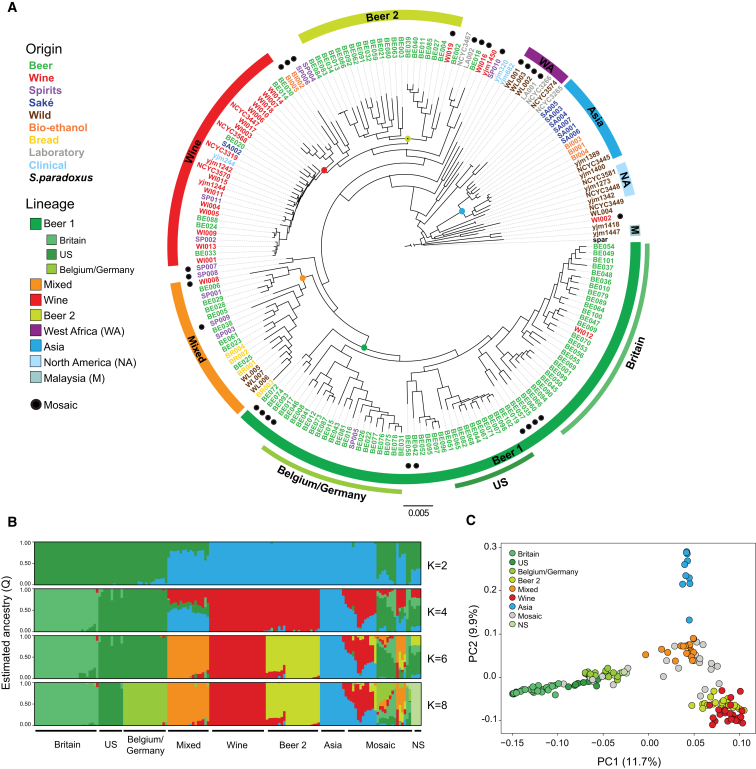
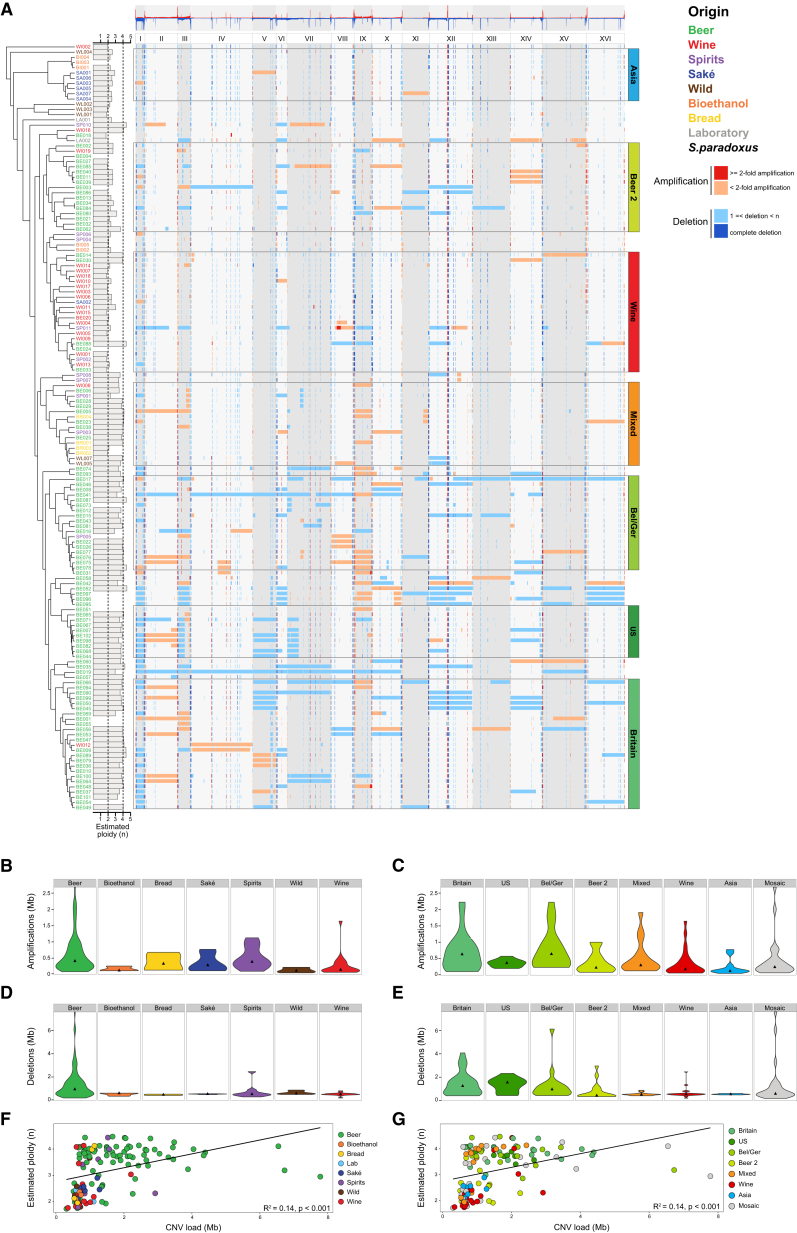
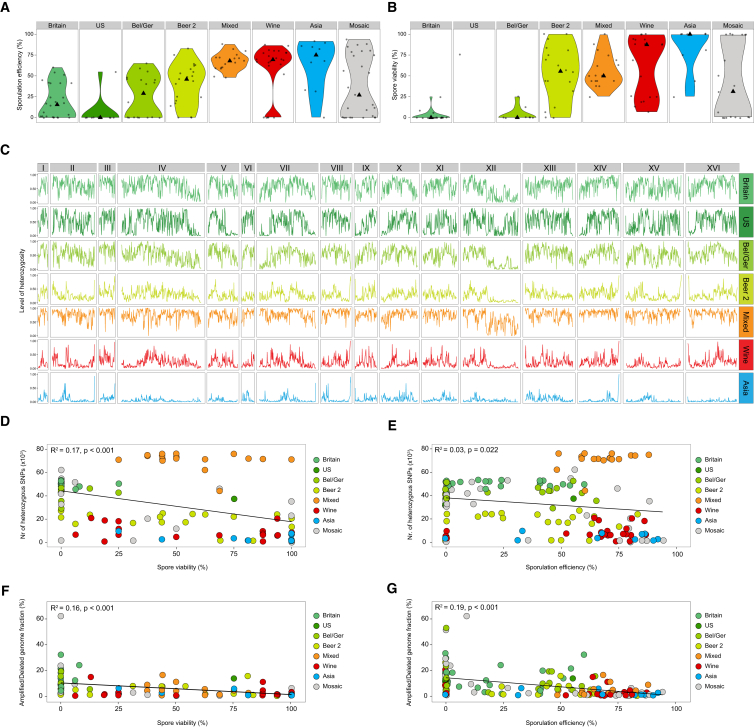
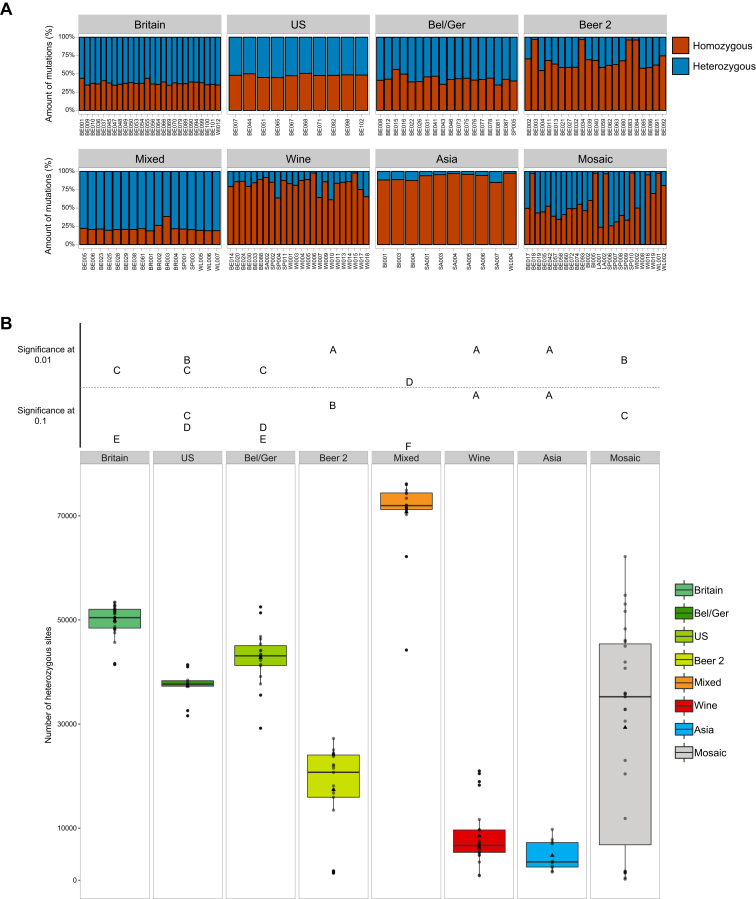
Whereas domestication of livestock, pets, and crops is well documented, it is still unclear to what extent microbes associated with the production of food have also undergone human selection and where the plethora of industrial strains originates from. Here, we present the genomes and phenomes of 157 industrial Saccharomyces cerevisiae yeasts. Our analyses reveal that today's industrial yeasts can be divided into five sublineages that are genetically and phenotypically separated from wild strains and originate from only a few ancestors through complex patterns of domestication and local divergence. Large-scale phenotyping and genome analysis further show strong industry-specific selection for stress tolerance, sugar utilization, and flavor production, while the sexual cycle and other phenotypes related to survival in nature show decay, particularly in beer yeasts. Together, these results shed light on the origins, evolutionary history, and phenotypic diversity of industrial yeasts and provide a resource for further selection of superior strains. PAPERCLIP.
Copyright © 2016 The Author(s). Published by Elsevier Inc. All rights reserved.
Figures

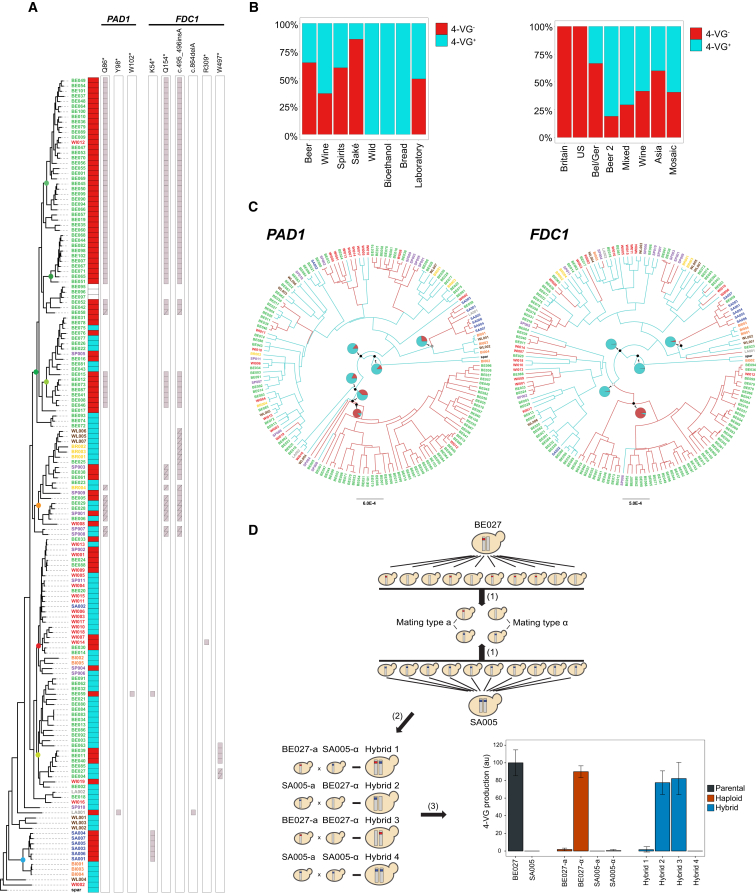
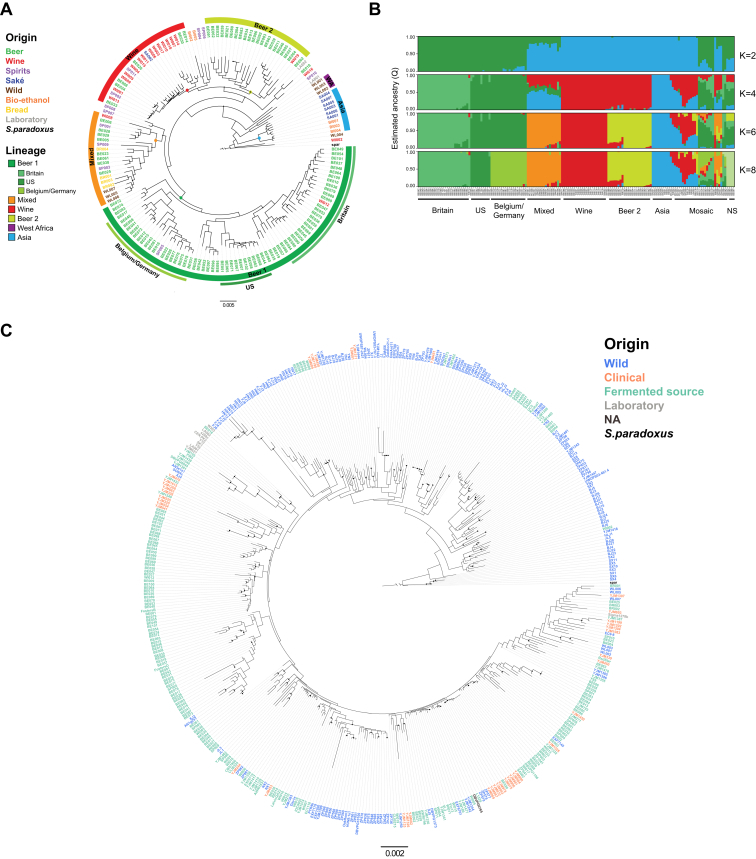
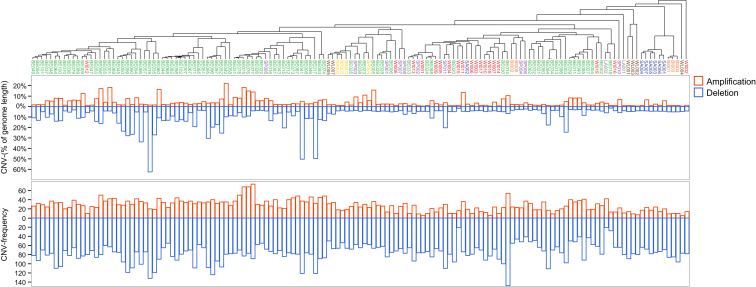
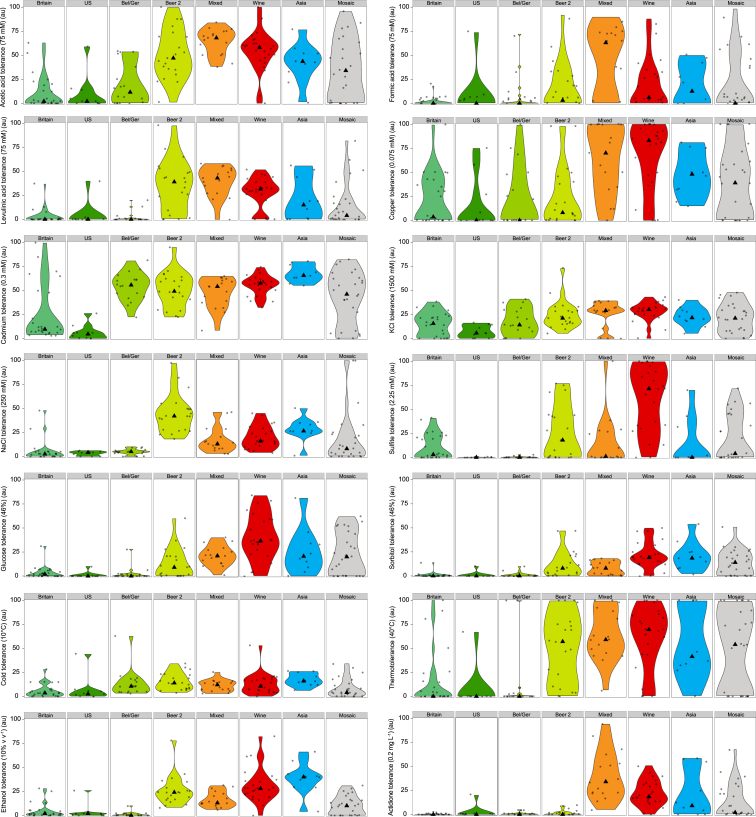
References
-
- Almeida P., Barbosa R., Zalar P., Imanishi Y., Shimizu K., Turchetti B., Legras J.L., Serra M., Dequin S., Couloux A. A population genomics insight into the Mediterranean origins of wine yeast domestication. Mol. Ecol. 2015;24:5412–5427. - PubMed
-
- Baele G., Lemey P. Bayesian evolutionary model testing in the phylogenomics era: matching model complexity with computational efficiency. Bioinformatics. 2013;29:1970–1979. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
