

GRIN2D Recurrent De Novo Dominant Mutation Causes a Severe Epileptic Encephalopathy Treatable with NMDA Receptor Channel Blockers
- PMID: 27616483
- PMCID: PMC5065652
- DOI: 10.1016/j.ajhg.2016.07.013
GRIN2D Recurrent De Novo Dominant Mutation Causes a Severe Epileptic Encephalopathy Treatable with NMDA Receptor Channel Blockers
Abstract
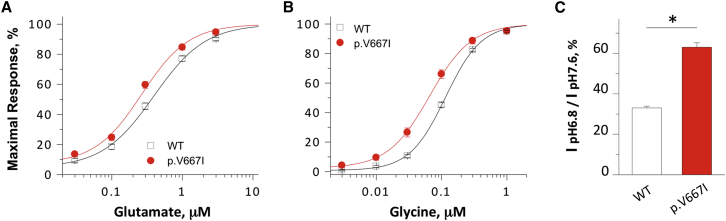
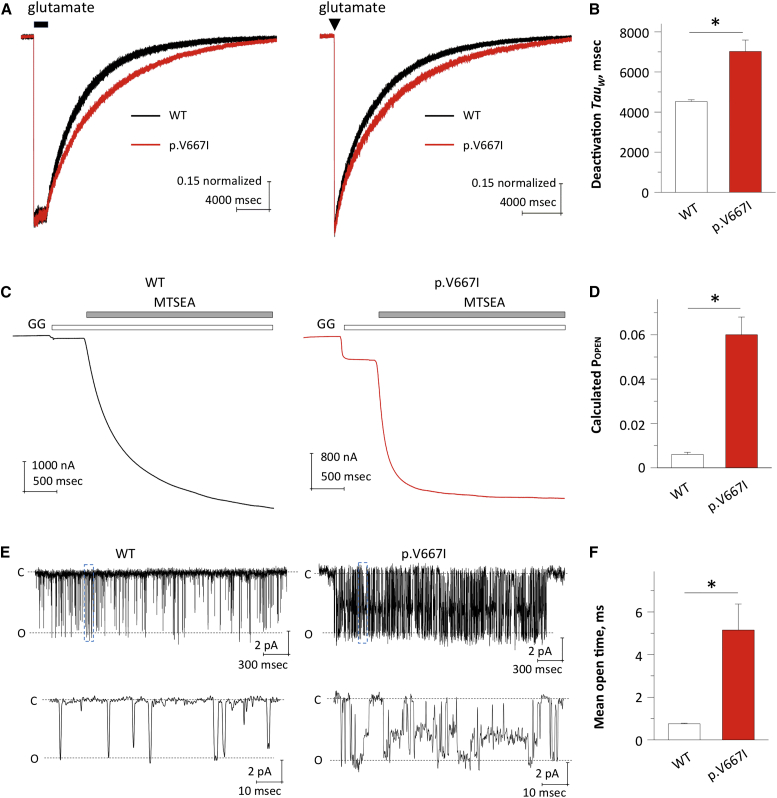
N-methyl-D-aspartate receptors (NMDARs) are ligand-gated cation channels that mediate excitatory synaptic transmission. Genetic mutations in multiple NMDAR subunits cause various childhood epilepsy syndromes. Here, we report a de novo recurrent heterozygous missense mutation-c.1999G>A (p.Val667Ile)-in a NMDAR gene previously unrecognized to harbor disease-causing mutations, GRIN2D, identified by exome and candidate panel sequencing in two unrelated children with epileptic encephalopathy. The resulting GluN2D p.Val667Ile exchange occurs in the M3 transmembrane domain involved in channel gating. This gain-of-function mutation increases glutamate and glycine potency by 2-fold, increases channel open probability by 6-fold, and reduces receptor sensitivity to endogenous negative modulators such as extracellular protons. Moreover, this mutation prolongs the deactivation time course after glutamate removal, which controls the synaptic time course. Transfection of cultured neurons with human GRIN2D cDNA harboring c.1999G>A leads to dendritic swelling and neuronal cell death, suggestive of excitotoxicity mediated by NMDAR over-activation. Because both individuals' seizures had proven refractory to conventional antiepileptic medications, the sensitivity of mutant NMDARs to FDA-approved NMDAR antagonists was evaluated. Based on these results, oral memantine was administered to both children, with resulting mild to moderate improvement in seizure burden and development. The older proband subsequently developed refractory status epilepticus, with dramatic electroclinical improvement upon treatment with ketamine and magnesium. Overall, these results suggest that NMDAR antagonists can be useful as adjuvant epilepsy therapy in individuals with GRIN2D gain-of-function mutations. This work further demonstrates the value of functionally evaluating a mutation, enabling mechanistic understanding and therapeutic modeling to realize precision medicine for epilepsy.
Copyright © 2016 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures




Comment in
-
Precision Medicine: NMDA Receptor-Targeted Therapy for GRIN2D Encephalopathy.Epilepsy Curr. 2017 Mar-Apr;17(2):112-114. doi: 10.5698/1535-7511.17.2.112. Epilepsy Curr. 2017. PMID: 28491004 Free PMC article. No abstract available.
References
-
- Srivastava S., Cohen J.S., Vernon H., Barañano K., McClellan R., Jamal L., Naidu S., Fatemi A. Clinical whole exome sequencing in child neurology practice. Ann. Neurol. 2014;76:473–483. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
