

Combined Analysis of Variation in Core, Accessory and Regulatory Genome Regions Provides a Super-Resolution View into the Evolution of Bacterial Populations
- PMID: 27618184
- PMCID: PMC5019451
- DOI: 10.1371/journal.pgen.1006280
Combined Analysis of Variation in Core, Accessory and Regulatory Genome Regions Provides a Super-Resolution View into the Evolution of Bacterial Populations
Abstract
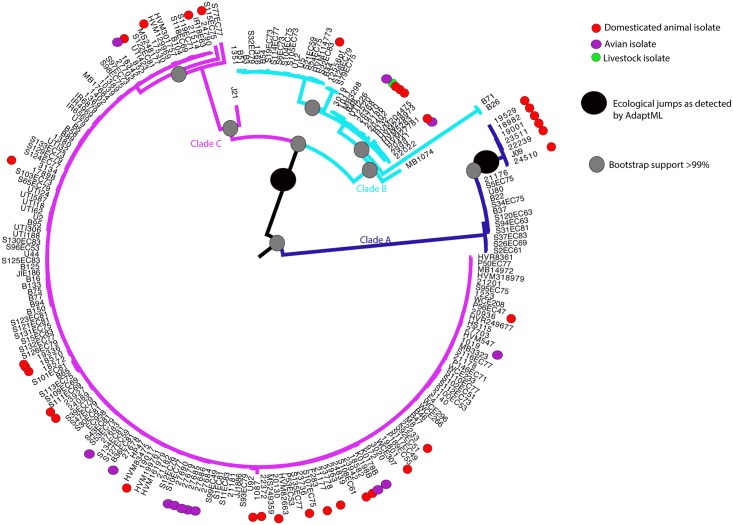
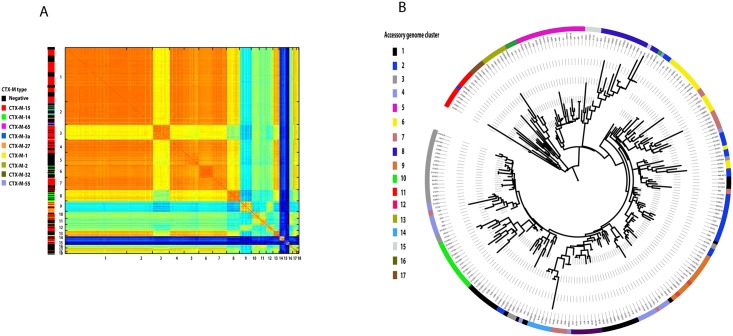
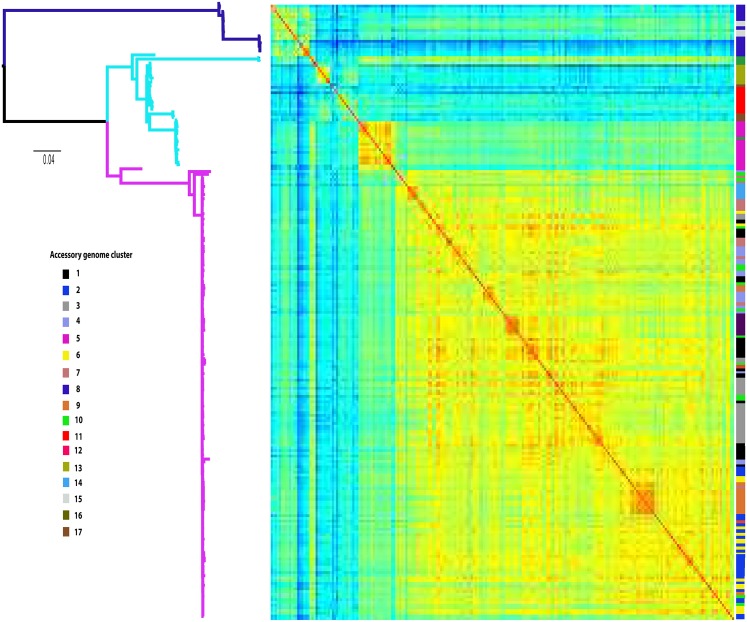
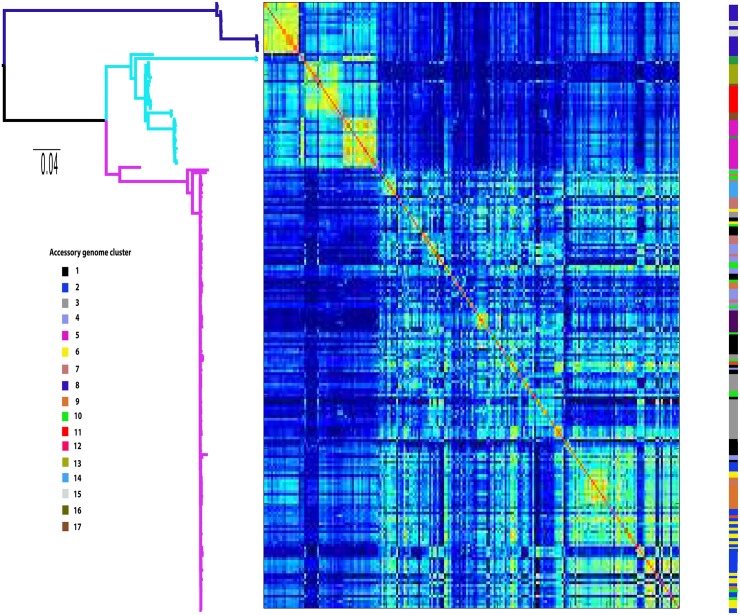
The use of whole-genome phylogenetic analysis has revolutionized our understanding of the evolution and spread of many important bacterial pathogens due to the high resolution view it provides. However, the majority of such analyses do not consider the potential role of accessory genes when inferring evolutionary trajectories. Moreover, the recently discovered importance of the switching of gene regulatory elements suggests that an exhaustive analysis, combining information from core and accessory genes with regulatory elements could provide unparalleled detail of the evolution of a bacterial population. Here we demonstrate this principle by applying it to a worldwide multi-host sample of the important pathogenic E. coli lineage ST131. Our approach reveals the existence of multiple circulating subtypes of the major drug-resistant clade of ST131 and provides the first ever population level evidence of core genome substitutions in gene regulatory regions associated with the acquisition and maintenance of different accessory genome elements.
Conflict of interest statement
The authors have declared no competing interests exist.
Figures
References
MeSH terms
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
