

Unsupervised detection of cancer driver mutations with parsimony-guided learning
- PMID: 27618449
- PMCID: PMC5328615
- DOI: 10.1038/ng.3658
Unsupervised detection of cancer driver mutations with parsimony-guided learning
Abstract
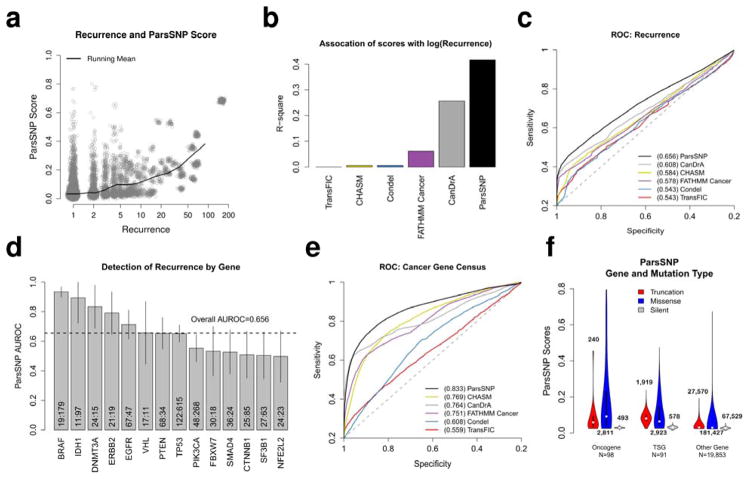
Methods are needed to reliably prioritize biologically active driver mutations over inactive passengers in high-throughput sequencing cancer data sets. We present ParsSNP, an unsupervised functional impact predictor that is guided by parsimony. ParsSNP uses an expectation-maximization framework to find mutations that explain tumor incidence broadly, without using predefined training labels that can introduce biases. We compare ParsSNP to five existing tools (CanDrA, CHASM, FATHMM Cancer, TransFIC, and Condel) across five distinct benchmarks. ParsSNP outperformed the existing tools in 24 of 25 comparisons. To investigate the real-world benefit of these improvements, we applied ParsSNP to an independent data set of 30 patients with diffuse-type gastric cancer. ParsSNP identified many known and likely driver mutations that other methods did not detect, including truncation mutations in known tumor suppressors and the recurrent driver substitution RHOA p.Tyr42Cys. In conclusion, ParsSNP uses an innovative, parsimony-based approach to prioritize cancer driver mutations and provides dramatic improvements over existing methods.
Conflict of interest statement
The authors declare no competing financial interests.
Figures



References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
