

High-throughput single-cell gene-expression profiling with multiplexed error-robust fluorescence in situ hybridization
- PMID: 27625426
- PMCID: PMC5047202
- DOI: 10.1073/pnas.1612826113
High-throughput single-cell gene-expression profiling with multiplexed error-robust fluorescence in situ hybridization
Abstract
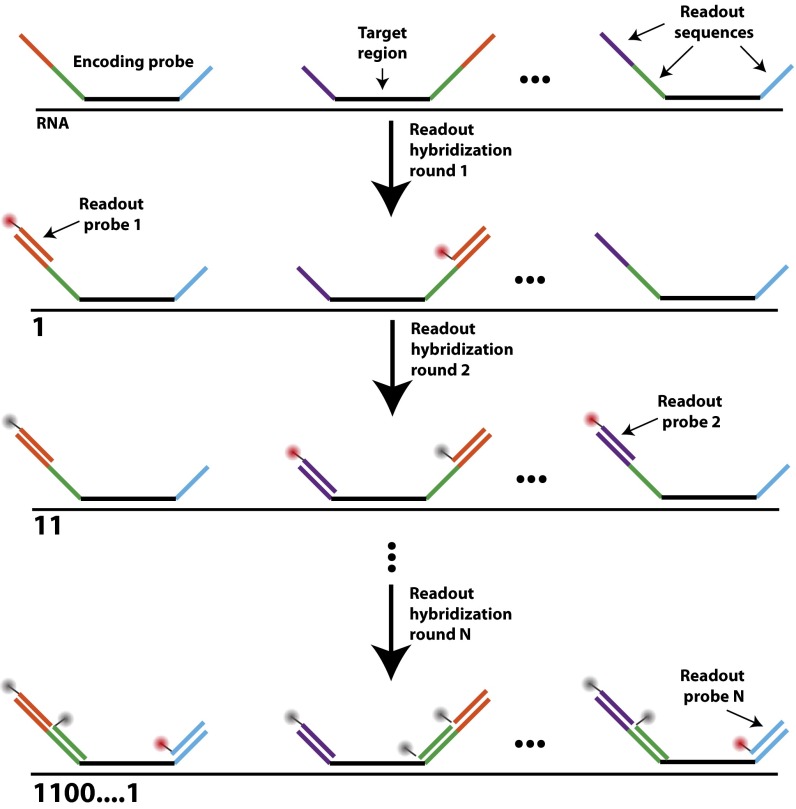
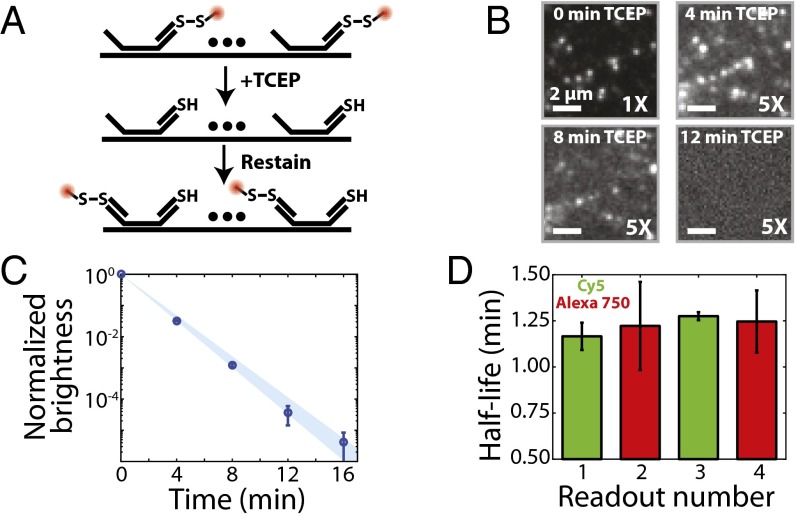
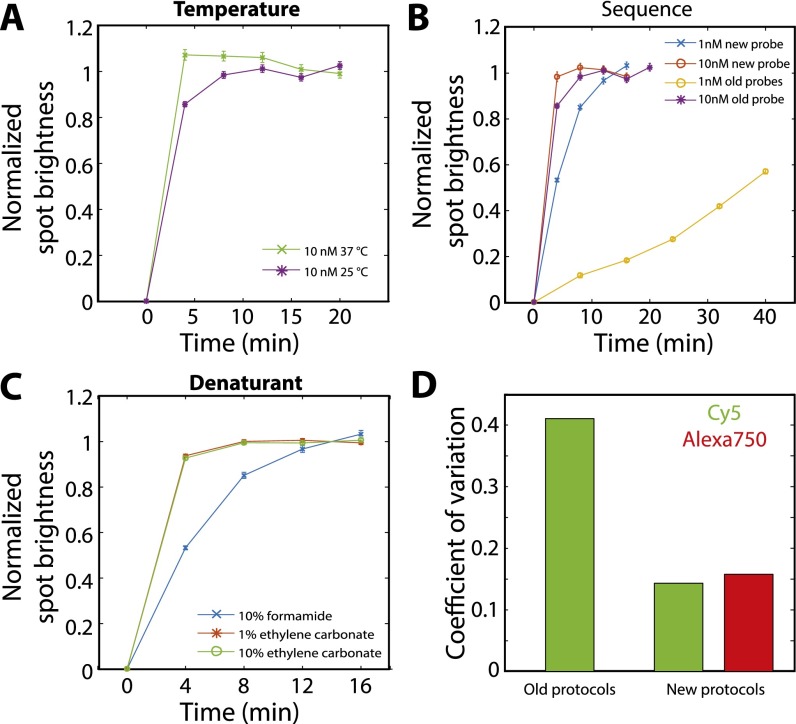
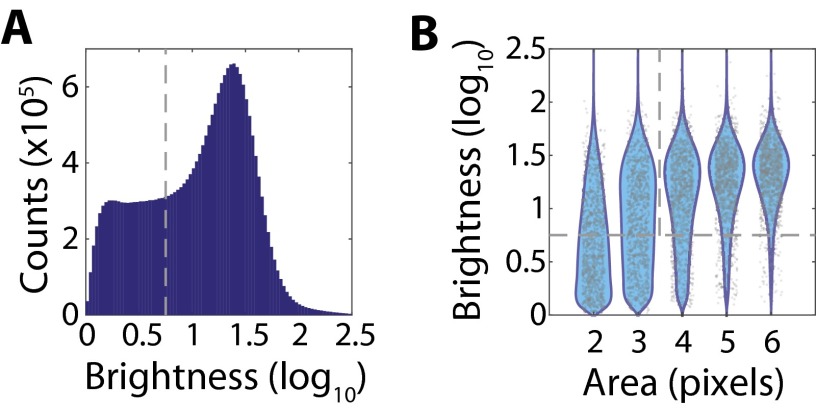
Image-based approaches to single-cell transcriptomics, in which RNA species are identified and counted in situ via imaging, have emerged as a powerful complement to single-cell methods based on RNA sequencing of dissociated cells. These image-based approaches naturally preserve the native spatial context of RNAs within a cell and the organization of cells within tissue, which are important for addressing many biological questions. However, the throughput of these image-based approaches is relatively low. Here we report advances that lead to a drastic increase in the measurement throughput of multiplexed error-robust fluorescence in situ hybridization (MERFISH), an image-based approach to single-cell transcriptomics. In MERFISH, RNAs are identified via a combinatorial labeling approach that encodes RNA species with error-robust barcodes followed by sequential rounds of single-molecule fluorescence in situ hybridization (smFISH) to read out these barcodes. Here we increase the throughput of MERFISH by two orders of magnitude through a combination of improvements, including using chemical cleavage instead of photobleaching to remove fluorescent signals between consecutive rounds of smFISH imaging, increasing the imaging field of view, and using multicolor imaging. With these improvements, we performed RNA profiling in more than 100,000 human cells, with as many as 40,000 cells measured in a single 18-h measurement. This throughput should substantially extend the range of biological questions that can be addressed by MERFISH.
Keywords: fluorescence; in situ hybridization; multiplexed imaging; single-cell analysis; transcriptomics.
Conflict of interest statement
X.Z., J.R.M., and K.H.C. are inventors on a patent applied for by Harvard University that covers the MERFISH method.
Figures


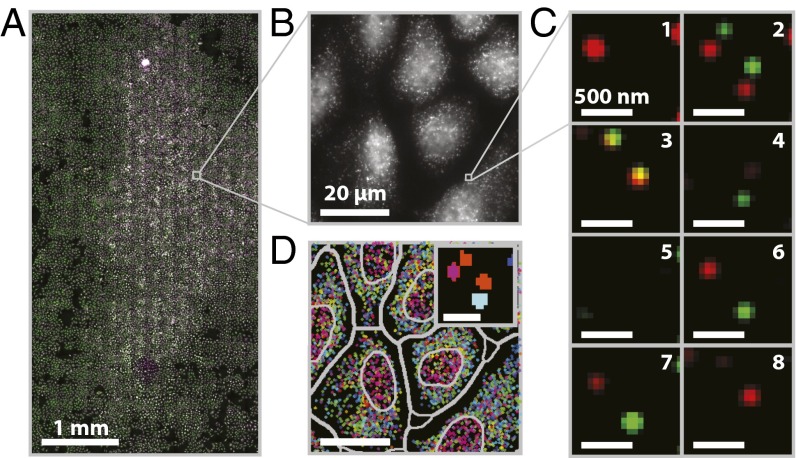
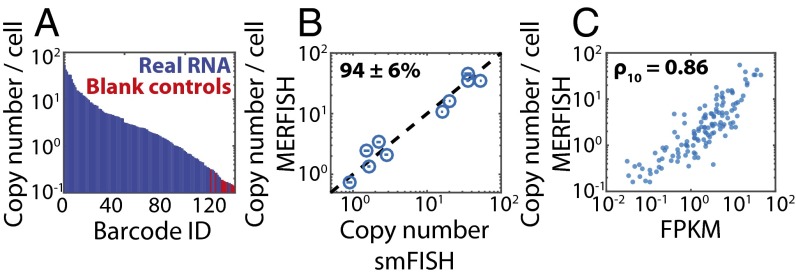
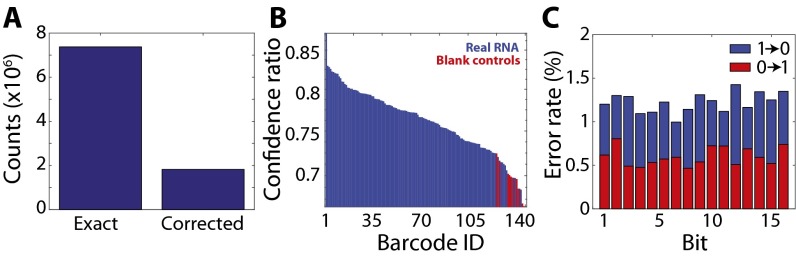
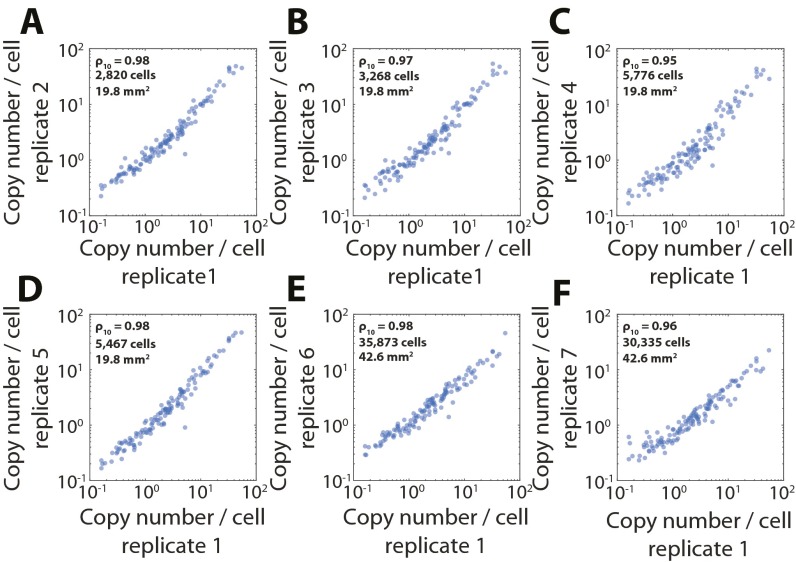
References
-
- Sandberg R. Entering the era of single-cell transcriptomics in biology and medicine. Nat Methods. 2014;11(1):22–24. - PubMed
-
- Eberwine J, Sul J-Y, Bartfai T, Kim J. The promise of single-cell sequencing. Nat Methods. 2014;11(1):25–27. - PubMed
-
- Shapiro E, Biezuner T, Linnarsson S. Single-cell sequencing-based technologies will revolutionize whole-organism science. Nat Rev Genet. 2013;14(9):618–630. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
