

Reactive Oxygen Species: Physiological and Physiopathological Effects on Synaptic Plasticity
- PMID: 27625575
- PMCID: PMC5012454
- DOI: 10.4137/JEN.S39887
Reactive Oxygen Species: Physiological and Physiopathological Effects on Synaptic Plasticity
Abstract
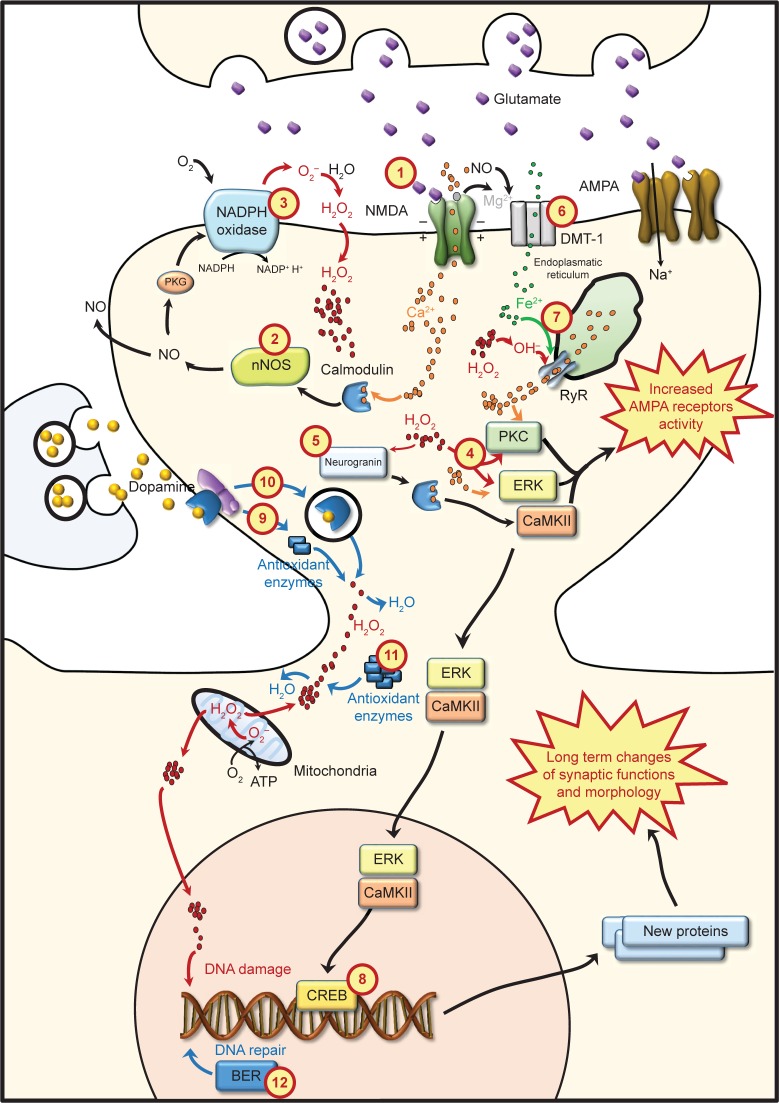
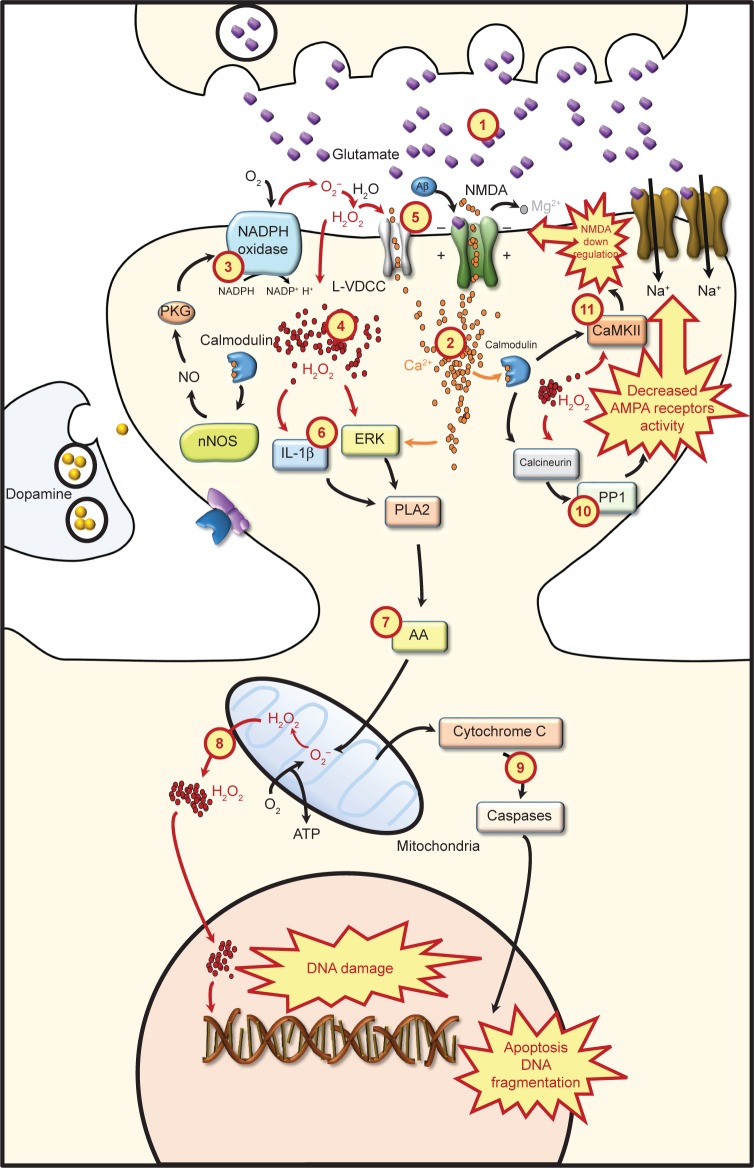
In the mammalian central nervous system, reactive oxygen species (ROS) generation is counterbalanced by antioxidant defenses. When large amounts of ROS accumulate, antioxidant mechanisms become overwhelmed and oxidative cellular stress may occur. Therefore, ROS are typically characterized as toxic molecules, oxidizing membrane lipids, changing the conformation of proteins, damaging nucleic acids, and causing deficits in synaptic plasticity. High ROS concentrations are associated with a decline in cognitive functions, as observed in some neurodegenerative disorders and age-dependent decay of neuroplasticity. Nevertheless, controlled ROS production provides the optimal redox state for the activation of transductional pathways involved in synaptic changes. Since ROS may regulate neuronal activity and elicit negative effects at the same time, the distinction between beneficial and deleterious consequences is unclear. In this regard, this review assesses current research and describes the main sources of ROS in neurons, specifying their involvement in synaptic plasticity and distinguishing between physiological and pathological processes implicated.
Keywords: oxidative stress; reactive oxygen species; synaptic plasticity.
Figures
References
-
- Elgersma Y, Silva AJ. Molecular mechanisms of synaptic plasticity and memory. Curr Opin Neurobiol. 1999;9(2):209–213. - PubMed
-
- Lynch MA. Long-term potentiation and memory. Physiol Rev. 2004;84(1):87–136. - PubMed
-
- Hidalgo C, Arias-Cavieres A. Calcium, reactive oxygen species, and synaptic plasticity. Physiology (Bethesda) 2016;31(3):201–215. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
