

Ibrutinib targets mutant-EGFR kinase with a distinct binding conformation
- PMID: 27626175
- PMCID: PMC5342513
- DOI: 10.18632/oncotarget.11951
Ibrutinib targets mutant-EGFR kinase with a distinct binding conformation
Abstract
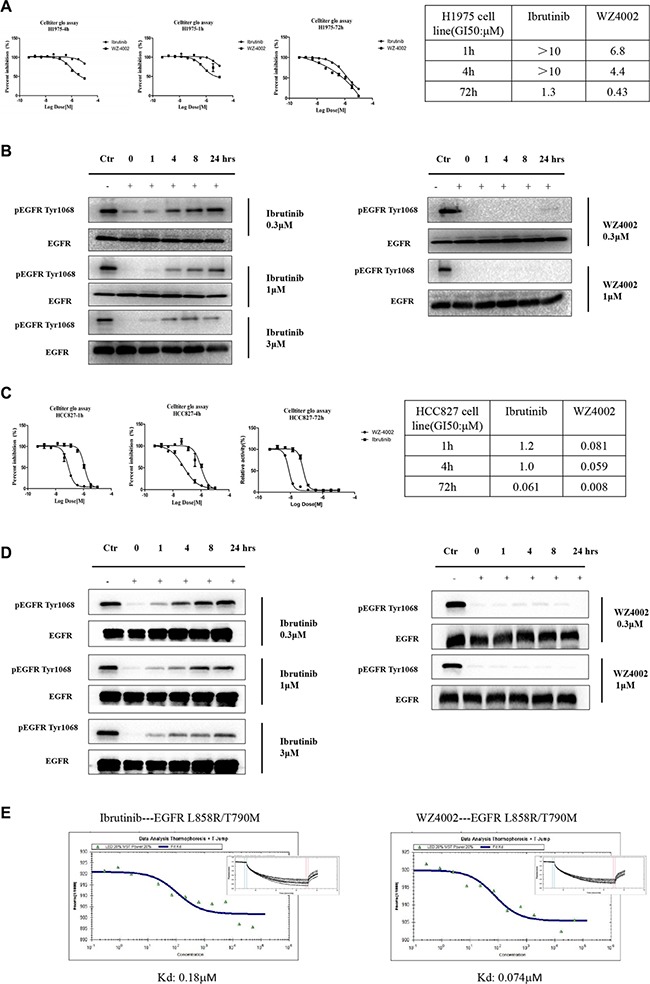
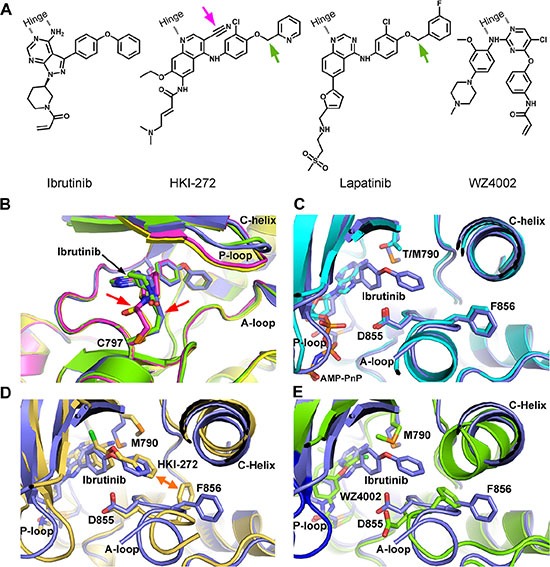
Ibrutinib, a clinically approved irreversible BTK kinase inhibitor for Mantle Cell Lymphoma (MCL) and Chronic Lymphocytic Leukemia (CLL) etc, has been reported to be potent against EGFR mutant kinase and currently being evaluated in clinic for Non Small Cell Lung Cancer (NSCLC). Through EGFR wt/mutant engineered isogenic BaF3 cell lines we confirmed the irreversible binding mode of Ibrutinib with EGFR wt/mutant kinase via Cys797. However, comparing to typical irreversible EGFR inhibitor, such as WZ4002, the washing-out experiments revealed a much less efficient covalent binding for Ibrutinib. The biochemical binding affinity examination in the EGFR L858R/T790M kinase revealed that, comparing to more efficient irreversible inhibitor WZ4002 (Kd: 0.074 μM), Ibrutinib exhibited less efficient binding (Kd: 0.18 μM). An X-ray crystal structure of EGFR (T790M) in complex with Ibrutinib exhibited a unique DFG-in/c-Helix-out inactive binding conformation, which partially explained the less efficiency of covalent binding and provided insight for further development of highly efficient irreversible binding inhibitor for the EGFR mutant kinase. These results also imply that, unlike the canonical irreversible inhibitor, sustained effective concentration might be required for Ibrutinib in order to achieve the maximal efficacy in the clinic application against EGFR driven NSCLC.
Keywords: DFG-in/c-Helix-out; EGFR kinase; Ibrutinib; NSCLC; inactive conformation.
Conflict of interest statement
Authors declare no conflicts of interest.
Figures
References
-
- Pan Z, Scheerens H, Li SJ, Schultz BE, Sprengeler PA, Burrill LC, Mendonca RV, Sweeney MD, Scott KC, Grothaus PG, Jeffery DA, Spoerke JM, Honigberg LA, et al. Discovery of selective irreversible inhibitors for Bruton's tyrosine kinase. Chem Med Chem. 2007;2:58–61. - PubMed
-
- Hendriks RW, Yuvaraj S, Kil LP. Targeting Bruton's tyrosine kinase in B cell malignancies. Nat Rev Cancer. 2014;14:219–232. - PubMed
-
- Herrera AF, Jacobsen ED. Ibrutinib for the treatment of mantle cell lymphoma. Clin Cancer Res. 2014;20:5365–5371. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
