

Accurate de novo design of hyperstable constrained peptides
- PMID: 27626386
- PMCID: PMC5161715
- DOI: 10.1038/nature19791
Accurate de novo design of hyperstable constrained peptides
Abstract
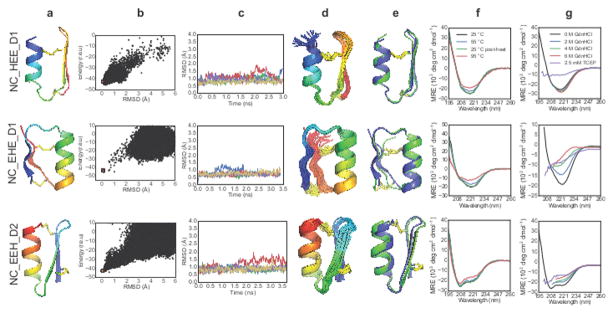
Naturally occurring, pharmacologically active peptides constrained with covalent crosslinks generally have shapes that have evolved to fit precisely into binding pockets on their targets. Such peptides can have excellent pharmaceutical properties, combining the stability and tissue penetration of small-molecule drugs with the specificity of much larger protein therapeutics. The ability to design constrained peptides with precisely specified tertiary structures would enable the design of shape-complementary inhibitors of arbitrary targets. Here we describe the development of computational methods for accurate de novo design of conformationally restricted peptides, and the use of these methods to design 18-47 residue, disulfide-crosslinked peptides, a subset of which are heterochiral and/or N-C backbone-cyclized. Both genetically encodable and non-canonical peptides are exceptionally stable to thermal and chemical denaturation, and 12 experimentally determined X-ray and NMR structures are nearly identical to the computational design models. The computational design methods and stable scaffolds presented here provide the basis for development of a new generation of peptide-based drugs.
Conflict of interest statement
Authors declare no competing financial interests.
Figures











References
-
- Conibear AC, et al. Approaches to the stabilization of bioactive epitopes by grafting and peptide cyclization. Biopolymers. 2016;106:89–100. - PubMed
-
- Craik DJ, Fairlie DP, Liras S, Price D. The future of peptide-based drugs. Chem Biol Drug Des. 2013;81:136–147. - PubMed
-
- Góngora-Benítez M, Tulla-Puche J, Albericio F. Multifaceted roles of disulfide bonds. Peptides as therapeutics. Chem Rev. 2014;114:901–926. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
