

Galectin-3 regulates inflammasome activation in cholestatic liver injury
- PMID: 27630169
- PMCID: PMC5102125
- DOI: 10.1096/fj.201600392RR
Galectin-3 regulates inflammasome activation in cholestatic liver injury
Abstract
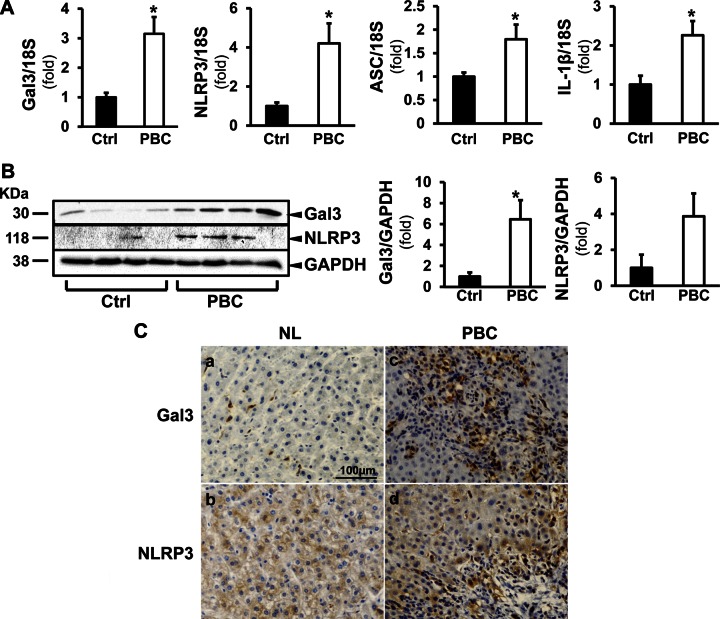
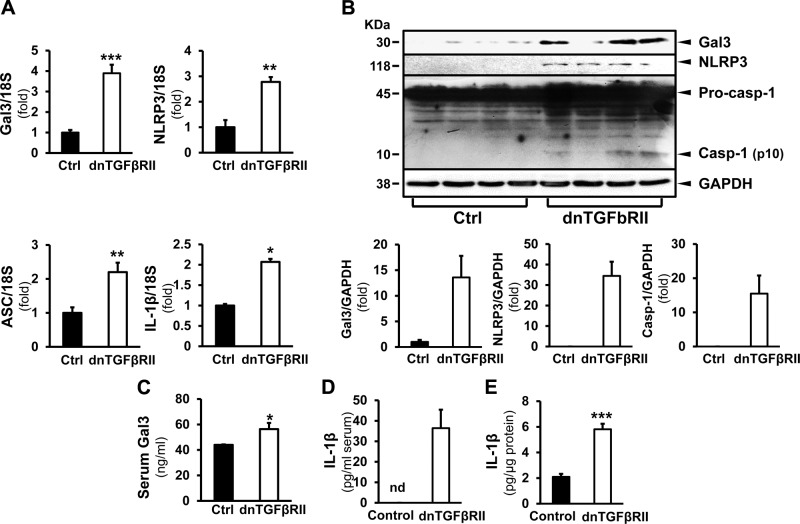
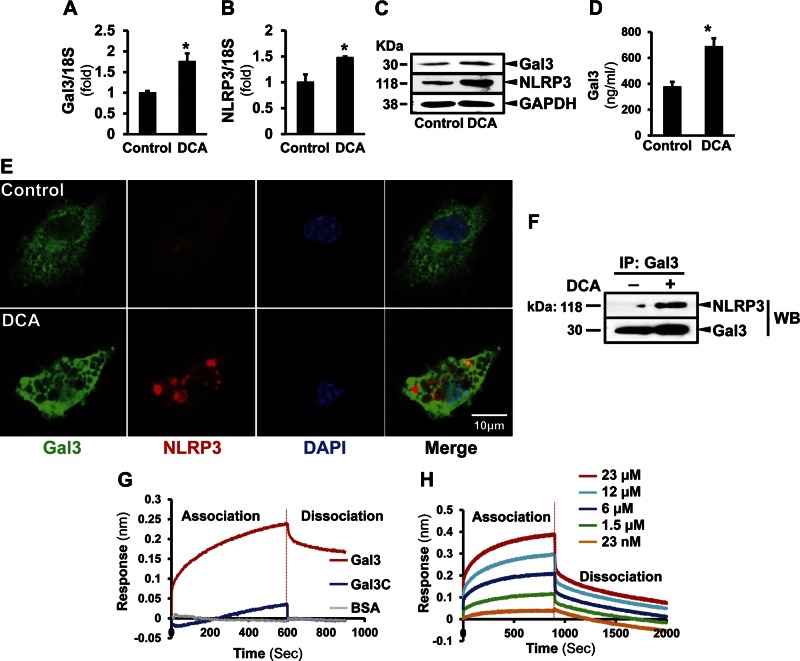
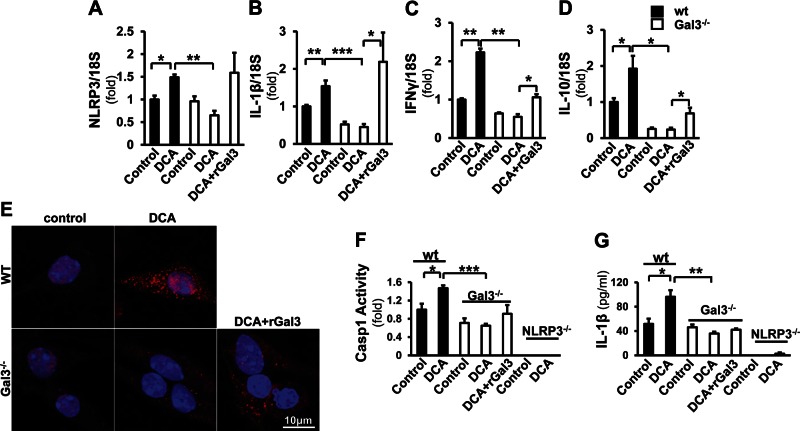
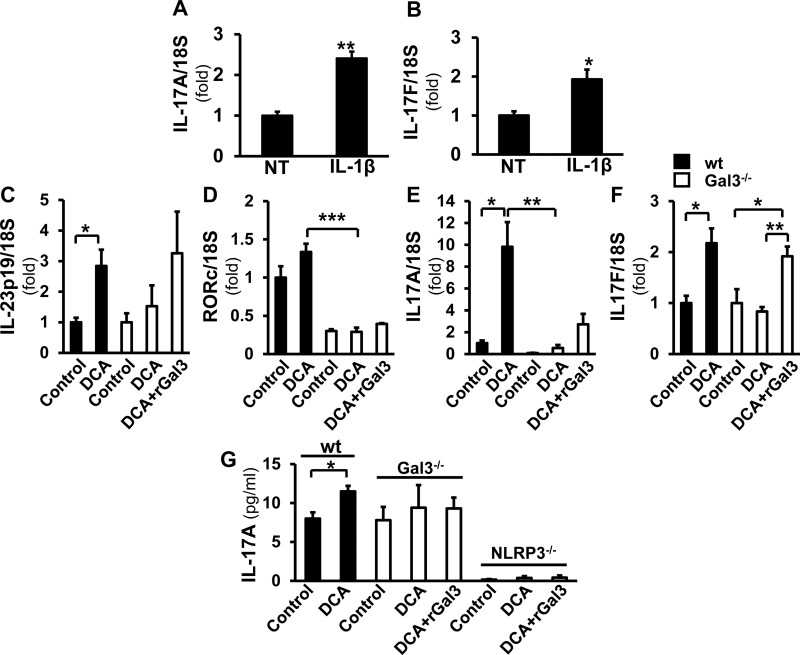
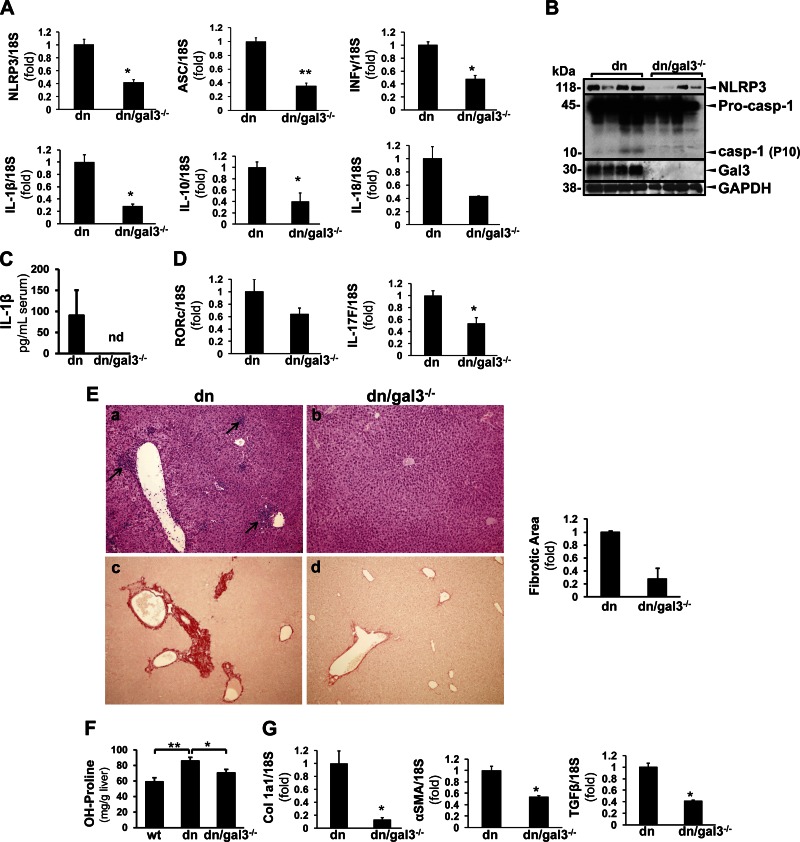
Macrophage activation is an important feature of primary biliary cholangitis (PBC) pathogenesis and other cholestatic liver diseases. Galectin-3 (Gal3), a pleiotropic lectin, is produced by monocytic cells and macrophages. However, its role in PBC has not been addressed. We hypothesized that Gal3 is a key to induce NOD-like receptor family, pyrin domain containing 3 (NLRP3) inflammasome in macrophages and in turn to propagate proinflammatory IL-17 signaling. In liver tissues from patients with PBC and dnTGF-βRII mice, a model of autoimmune cholangitis, the expression of Gal3, NLRP3, and the adaptor protein adaptor apoptosis-associated speck-like protein was induced, with the downstream activation of caspase-1 and IL-1β. In wild-type hepatic macrophages, deoxycholic acid induced the association of Gal3 and NLRP3 with direct activation of the inflammasome, resulting in an increase in IL-1β. Downstream retinoid-related orphan receptor C mRNA, IL-17A, and IL-17F were induced. In Gal3-/- macrophages, no inflammasome activation was detected. To confirm the key role of Gal3 in the pathogenesis of cholestatic liver injury, we generated dnTGF-βRII/galectin-3-/- (dn/Gal3-/-) mice, which showed impaired inflammasome activation along with significantly improved inflammation and fibrosis. Taken together, our data point to a novel role of Gal3 as an initiator of inflammatory signaling in autoimmune cholangitis, mediating the activation of NLRP3 inflammasome and inducing IL-17 proinflammatory cascades. These studies provide a rationale to target Gal3 in autoimmune cholangitis and potentially other cholestatic diseases.-Tian, J., Yang, G., Chen, H.-Y., Hsu, D. K., Tomilov, A., Olson, K. A., Dehnad, A., Fish, S. R., Cortopassi, G., Zhao, B., Liu, F.-T., Gershwin, M. E., Török, N. J., Jiang, J. X. Galectin-3 regulates inflammasome activation in cholestatic liver injury.
Keywords: IL-17; NLRP3; galectin-3; primary biliary cholangitis.
© FASEB.
Figures
References
-
- He X. S., Ansari A. A., Ridgway W. M., Coppel R. L., Gershwin M. E. (2006) New insights to the immunopathology and autoimmune responses in primary biliary cirrhosis. Cell. Immunol. 239, 1–13 - PubMed
-
- Chuang Y. H., Lian Z. X., Yang G. X., Shu S. A., Moritoki Y., Ridgway W. M., Ansari A. A., Kronenberg M., Flavell R. A., Gao B., Gershwin M. E. (2008) Natural killer T cells exacerbate liver injury in a transforming growth factor beta receptor II dominant-negative mouse model of primary biliary cirrhosis. Hepatology 47, 571–580 - PubMed
-
- Lleo A., Bowlus C. L., Yang G. X., Invernizzi P., Podda M., Van de Water J., Ansari A. A., Coppel R. L., Worman H. J., Gores G. J., Gershwin M. E. (2010) Biliary apotopes and anti-mitochondrial antibodies activate innate immune responses in primary biliary cirrhosis. Hepatology 52, 987–998 - PMC - PubMed
-
- Jiang J. X., Chen X., Hsu D. K., Baghy K., Serizawa N., Scott F., Takada Y., Takada Y., Fukada H., Chen J., Devaraj S., Adamson R., Liu F.-T., Török N. J. (2012) Galectin-3 modulates phagocytosis-induced stellate cell activation and liver fibrosis in vivo. Am. J. Physiol. Gastrointest. Liver Physiol. 302, G439–G446 - PMC - PubMed
-
- Liu F.-T. (1990) Molecular biology of IgE-binding protein, IgE-binding factors, and IgE receptors. Crit. Rev. Immunol. 10, 289–306 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
