

Systems Biology-Based Investigation of Cellular Antiviral Drug Targets Identified by Gene-Trap Insertional Mutagenesis
- PMID: 27632082
- PMCID: PMC5025164
- DOI: 10.1371/journal.pcbi.1005074
Systems Biology-Based Investigation of Cellular Antiviral Drug Targets Identified by Gene-Trap Insertional Mutagenesis
Abstract
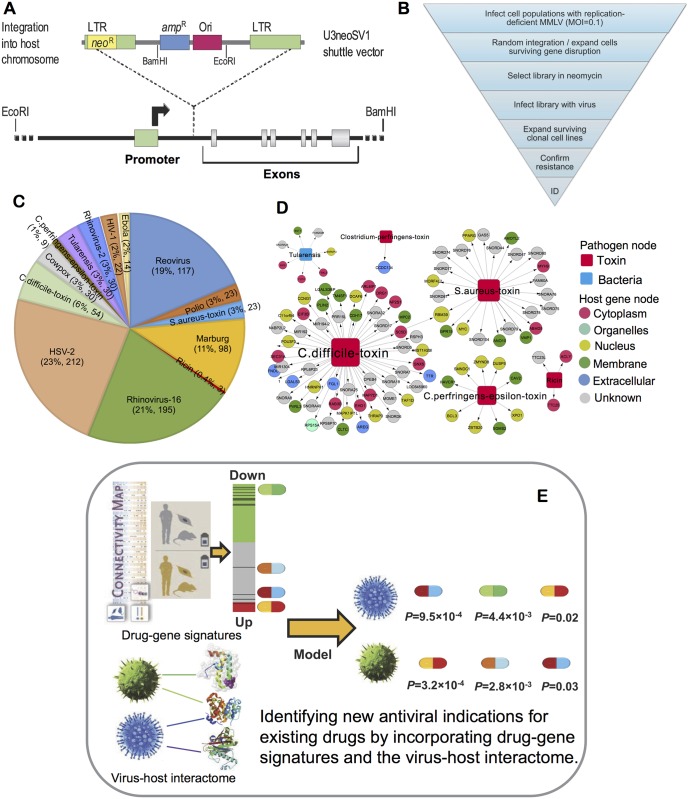
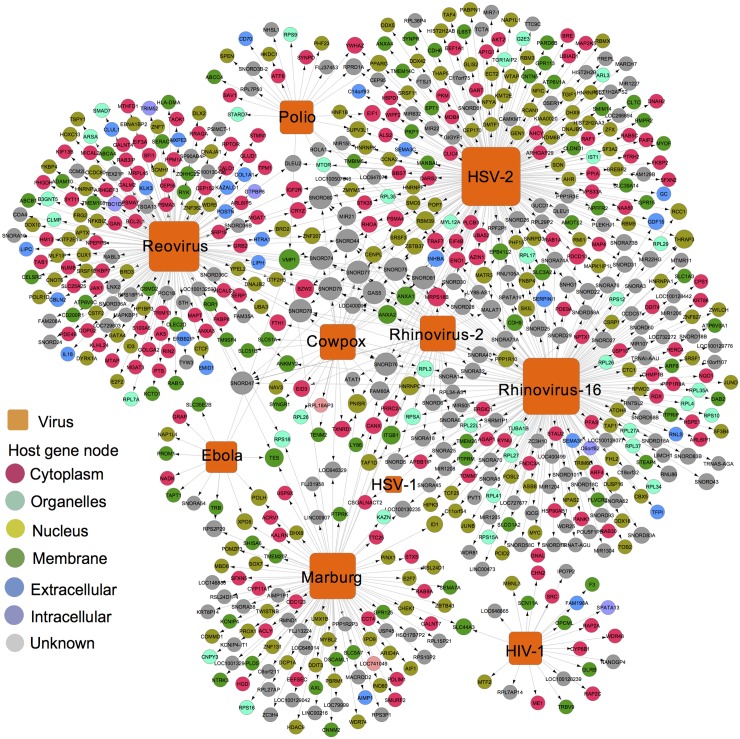
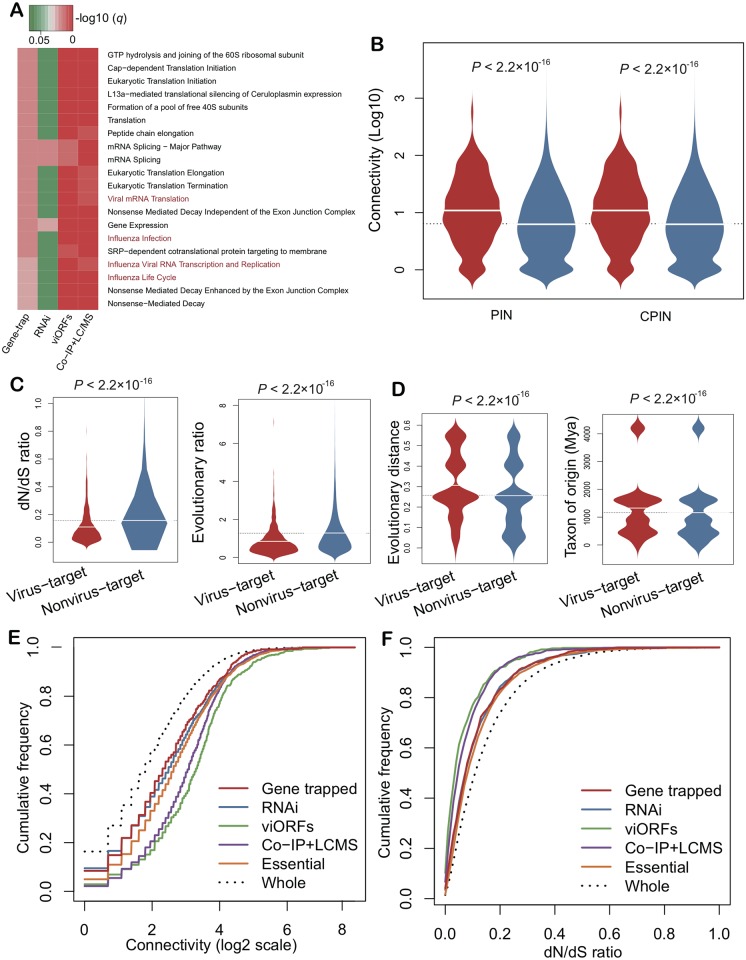
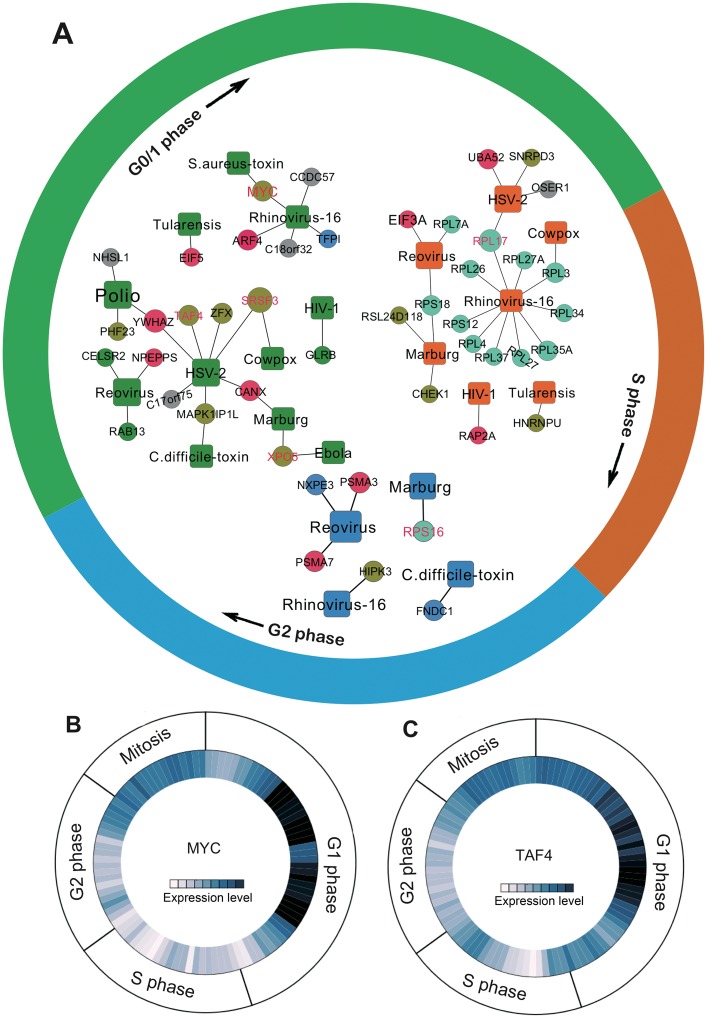
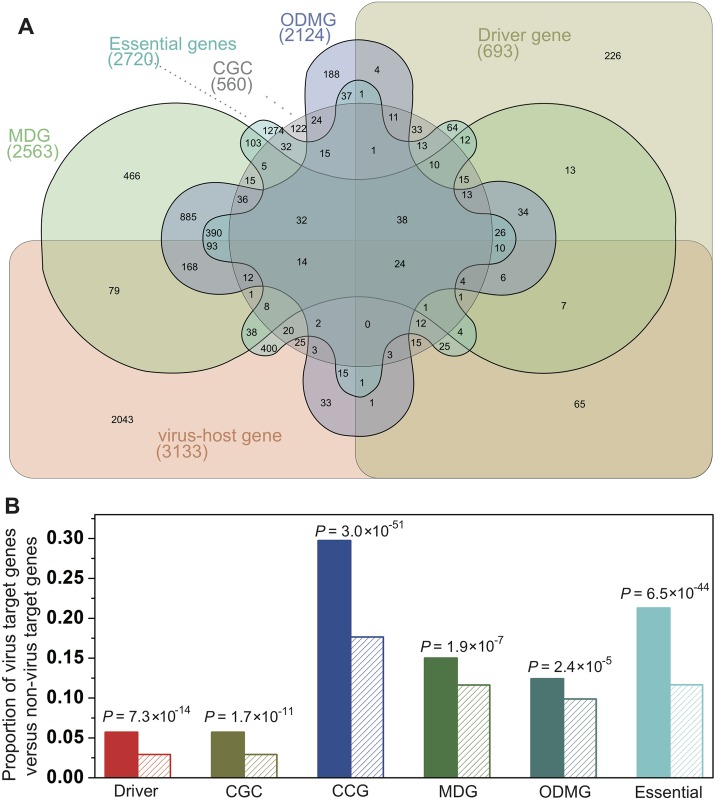
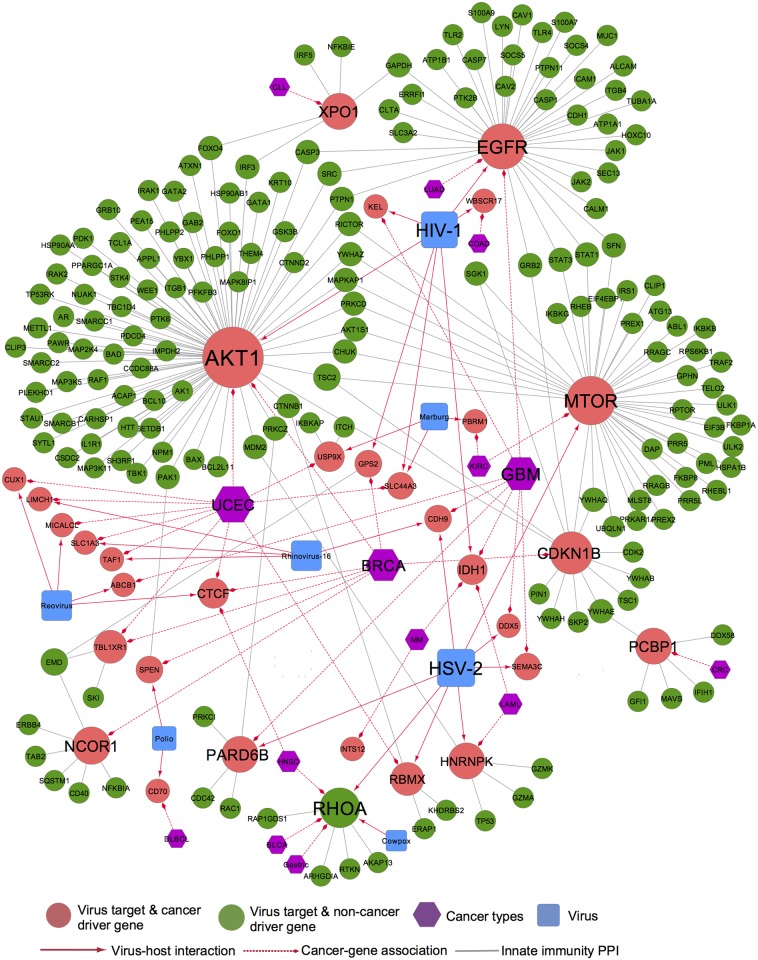
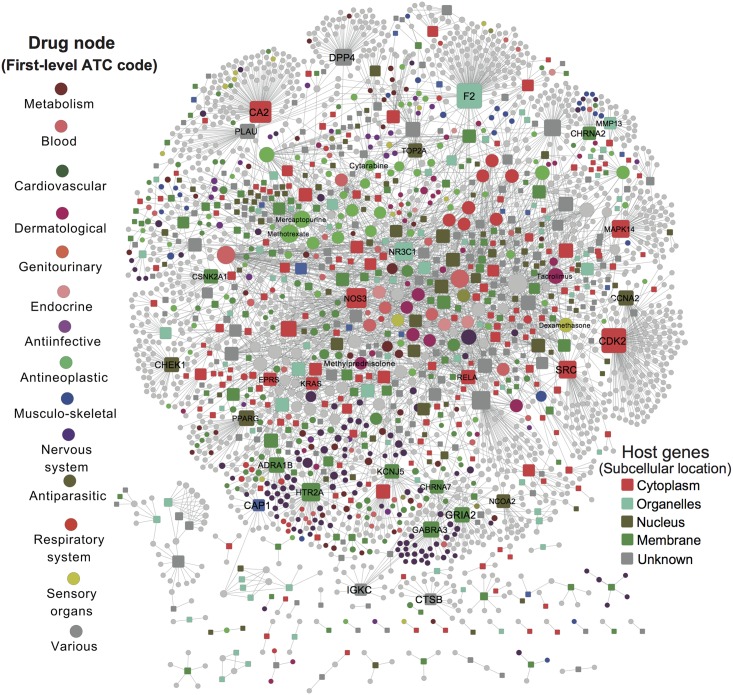
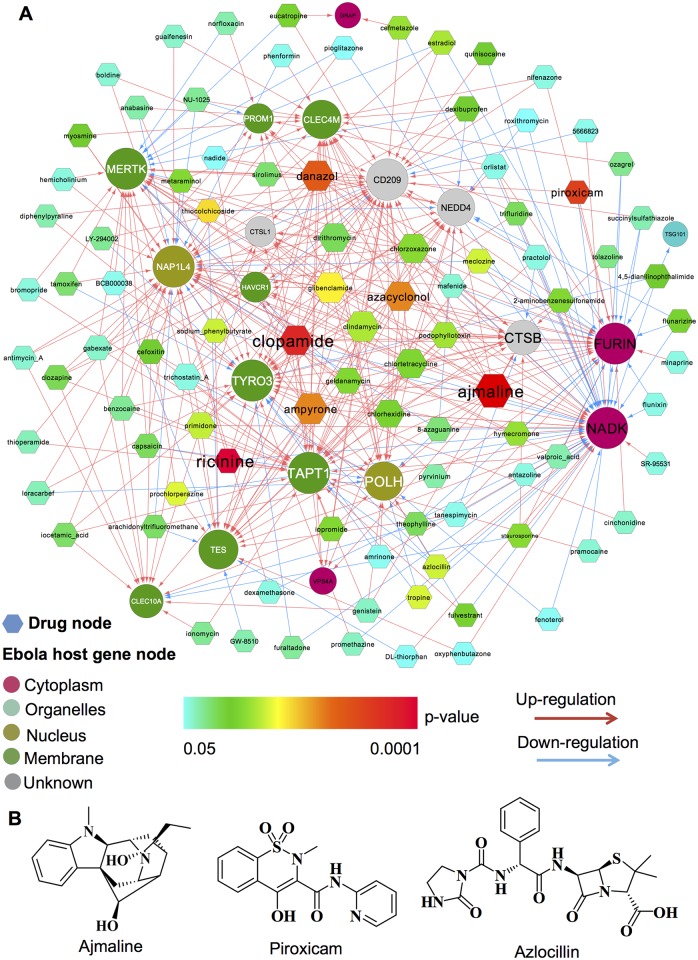
Viruses require host cellular factors for successful replication. A comprehensive systems-level investigation of the virus-host interactome is critical for understanding the roles of host factors with the end goal of discovering new druggable antiviral targets. Gene-trap insertional mutagenesis is a high-throughput forward genetics approach to randomly disrupt (trap) host genes and discover host genes that are essential for viral replication, but not for host cell survival. In this study, we used libraries of randomly mutagenized cells to discover cellular genes that are essential for the replication of 10 distinct cytotoxic mammalian viruses, 1 gram-negative bacterium, and 5 toxins. We herein reported 712 candidate cellular genes, characterizing distinct topological network and evolutionary signatures, and occupying central hubs in the human interactome. Cell cycle phase-specific network analysis showed that host cell cycle programs played critical roles during viral replication (e.g. MYC and TAF4 regulating G0/1 phase). Moreover, the viral perturbation of host cellular networks reflected disease etiology in that host genes (e.g. CTCF, RHOA, and CDKN1B) identified were frequently essential and significantly associated with Mendelian and orphan diseases, or somatic mutations in cancer. Computational drug repositioning framework via incorporating drug-gene signatures from the Connectivity Map into the virus-host interactome identified 110 putative druggable antiviral targets and prioritized several existing drugs (e.g. ajmaline) that may be potential for antiviral indication (e.g. anti-Ebola). In summary, this work provides a powerful methodology with a tight integration of gene-trap insertional mutagenesis testing and systems biology to identify new antiviral targets and drugs for the development of broadly acting and targeted clinical antiviral therapeutics.
Conflict of interest statement
JLM was previously employed by Zirus, Inc and is currently employed by GeneTAG Technology, Inc. The authors confirm they have no competing interests.
Figures
References
-
- 2013 Report on the Global AIDS Epidemic. Website: http://www.unaids.org/en/resources/campaigns/globalreport2013/factsheet/.
-
- 2014 Ebola Outbreak in West Africa-Reported Cases. Website: http://www.cdc.gov/vhf/ebola/outbreaks/2014-west-africa/cumulative-cases....
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
