

DNA replication stress mediates APOBEC3 family mutagenesis in breast cancer
- PMID: 27634334
- PMCID: PMC5025597
- DOI: 10.1186/s13059-016-1042-9
DNA replication stress mediates APOBEC3 family mutagenesis in breast cancer
Abstract
Background: The APOBEC3 family of cytidine deaminases mutate the cancer genome in a range of cancer types. Although many studies have documented the downstream effects of APOBEC3 activity through next-generation sequencing, less is known about their upstream regulation. In this study, we sought to identify a molecular basis for APOBEC3 expression and activation.
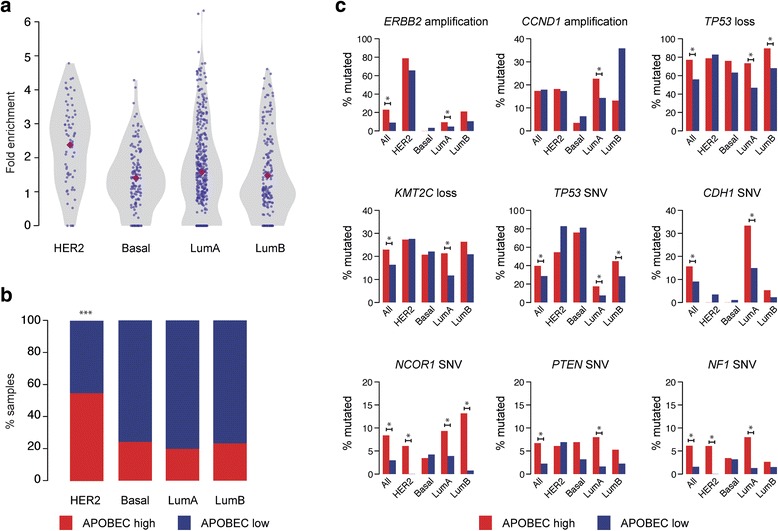
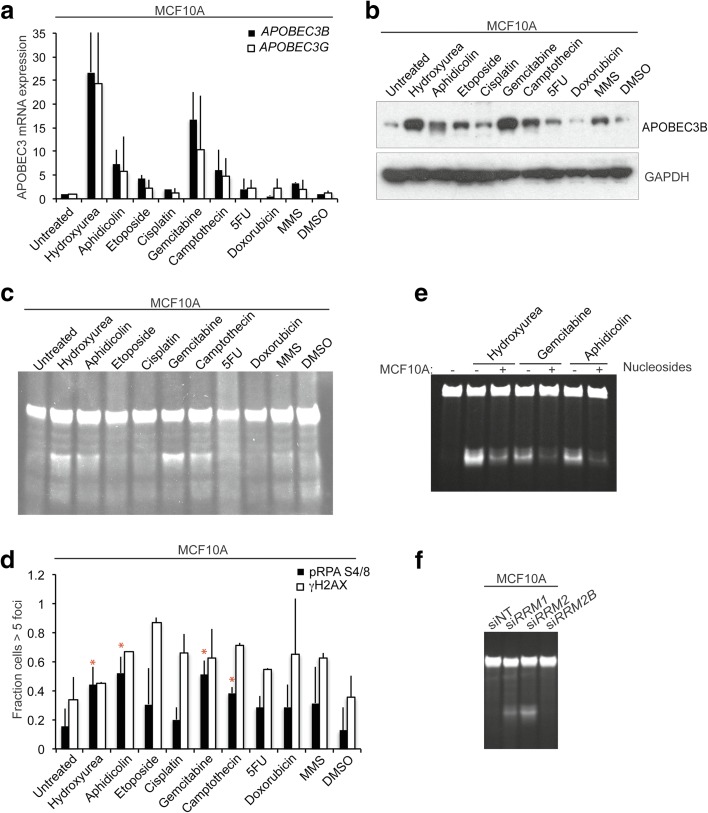
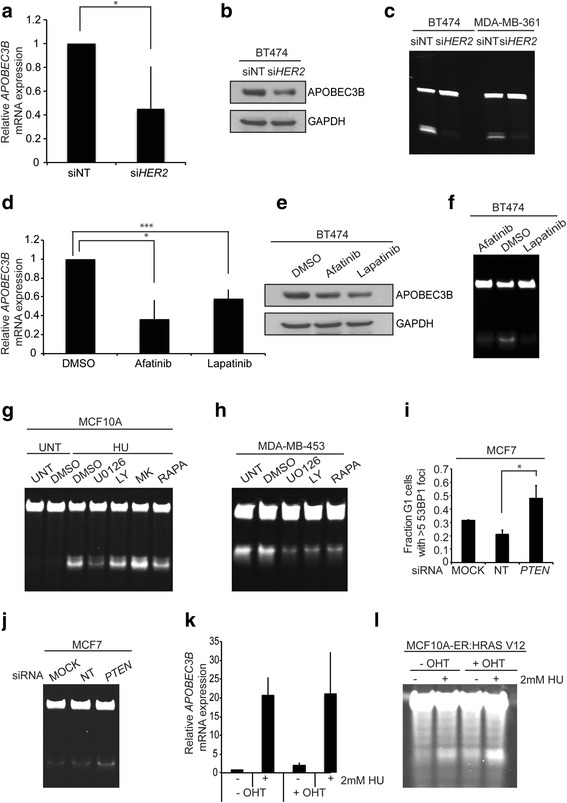
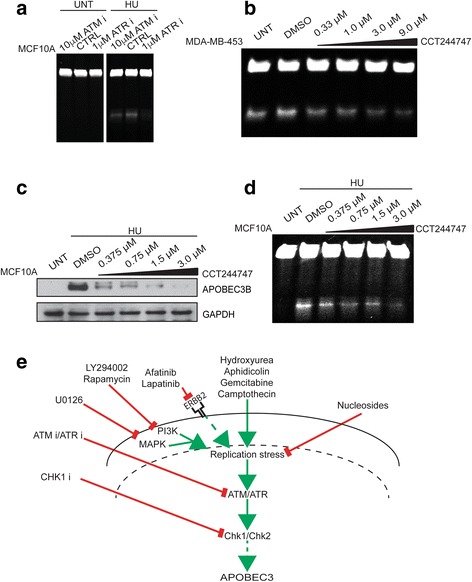
Results: HER2 amplification and PTEN loss promote DNA replication stress and APOBEC3B activity in vitro and correlate with APOBEC3 mutagenesis in vivo. HER2-enriched breast carcinomas display evidence of elevated levels of replication stress-associated DNA damage in vivo. Chemical and cytotoxic induction of replication stress, through aphidicolin, gemcitabine, camptothecin or hydroxyurea exposure, activates transcription of APOBEC3B via an ATR/Chk1-dependent pathway in vitro. APOBEC3B activation can be attenuated through repression of oncogenic signalling, small molecule inhibition of receptor tyrosine kinase signalling and alleviation of replication stress through nucleoside supplementation.
Conclusion: These data link oncogene, loss of tumour suppressor gene and drug-induced replication stress with APOBEC3B activity, providing new insights into how cytidine deaminase-induced mutagenesis might be activated in tumourigenesis and limited therapeutically.
Keywords: APOBEC; Genomic instability; Replication stress; Somatic mutation.
Figures

Comment in
-
DNA replication stress: a source of APOBEC3B expression in breast cancer.Genome Biol. 2016 Sep 30;17(1):202. doi: 10.1186/s13059-016-1069-y. Genome Biol. 2016. PMID: 27716362 Free PMC article.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
