

PHD3 Loss in Cancer Enables Metabolic Reliance on Fatty Acid Oxidation via Deactivation of ACC2
- PMID: 27635760
- PMCID: PMC5040345
- DOI: 10.1016/j.molcel.2016.08.014
PHD3 Loss in Cancer Enables Metabolic Reliance on Fatty Acid Oxidation via Deactivation of ACC2
Abstract
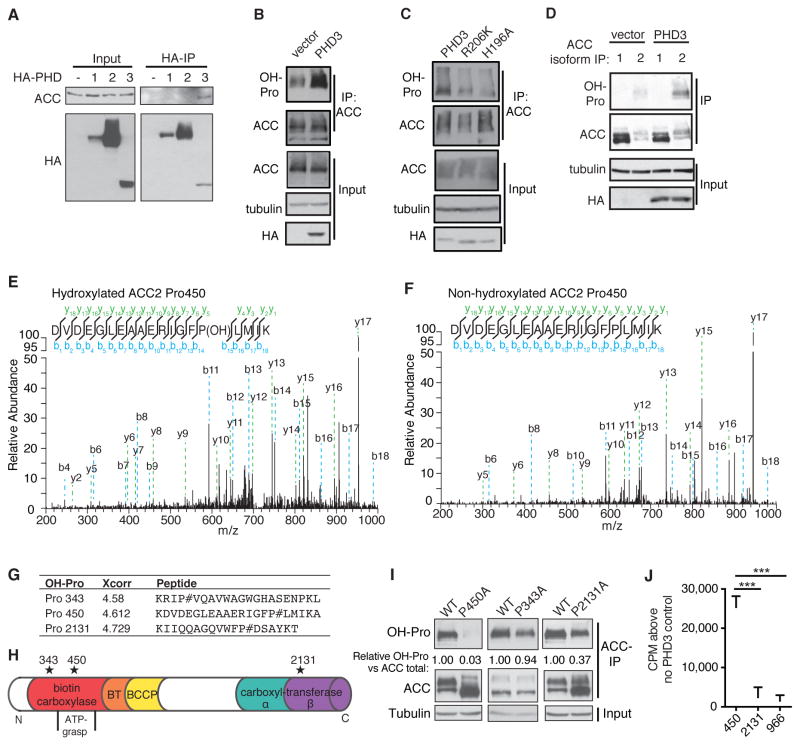
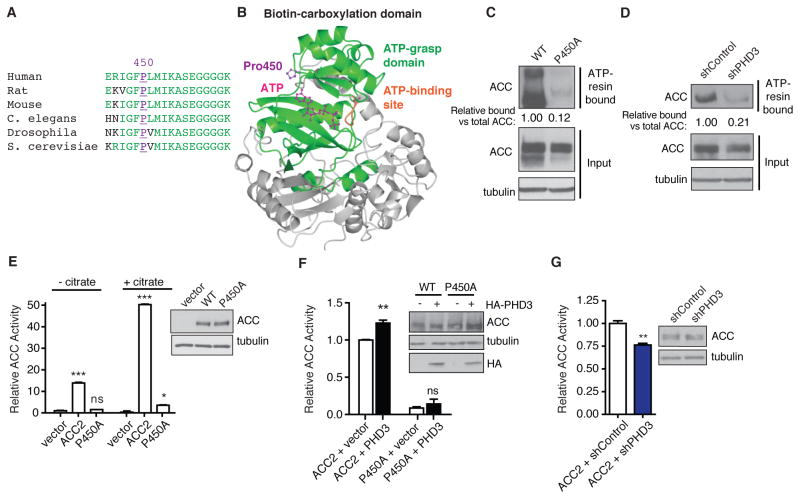
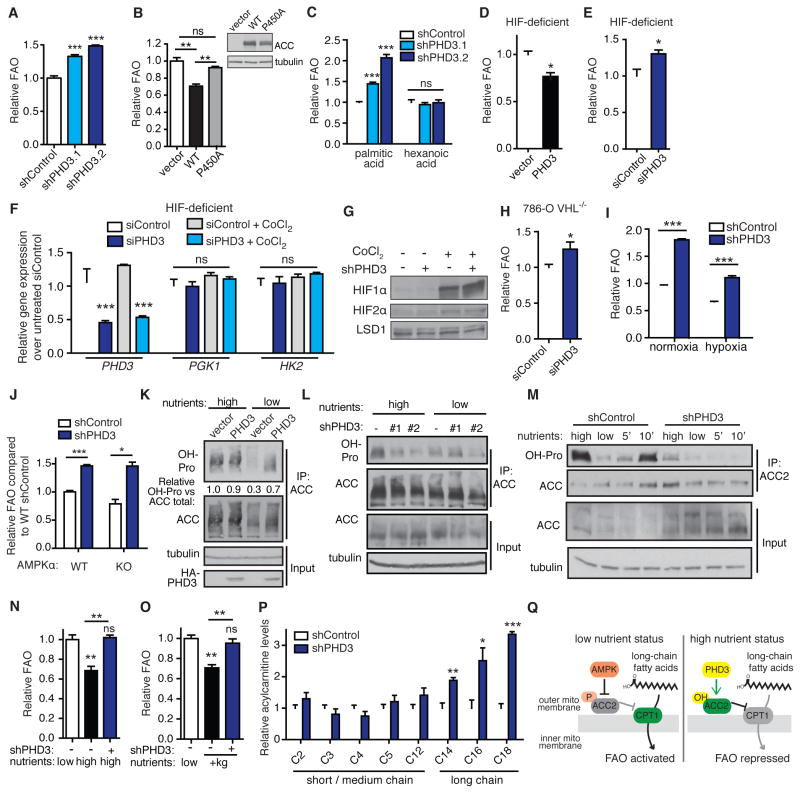
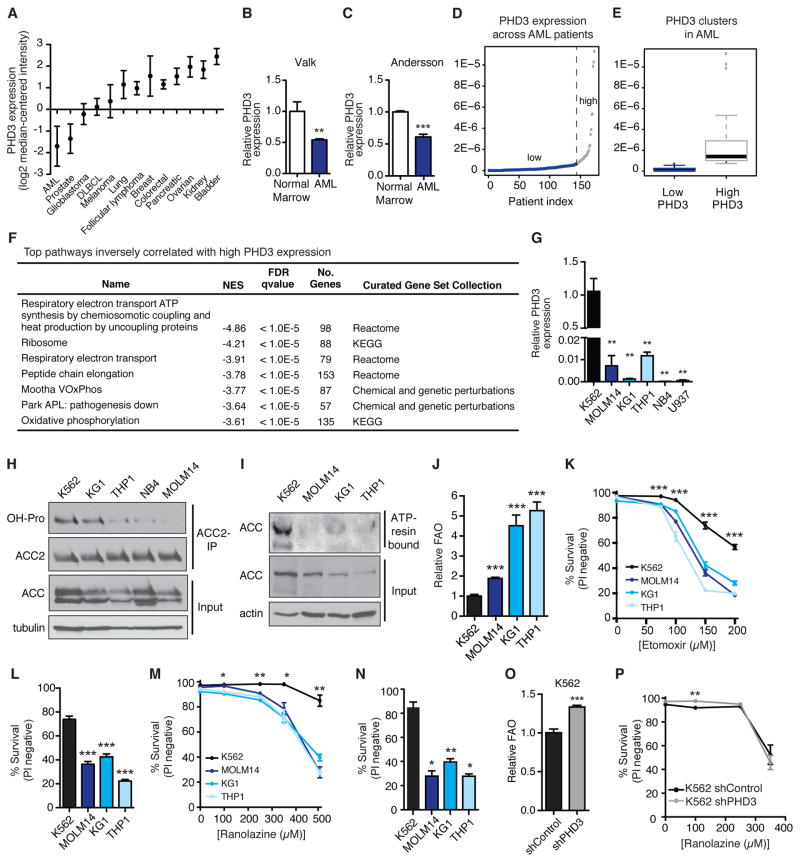
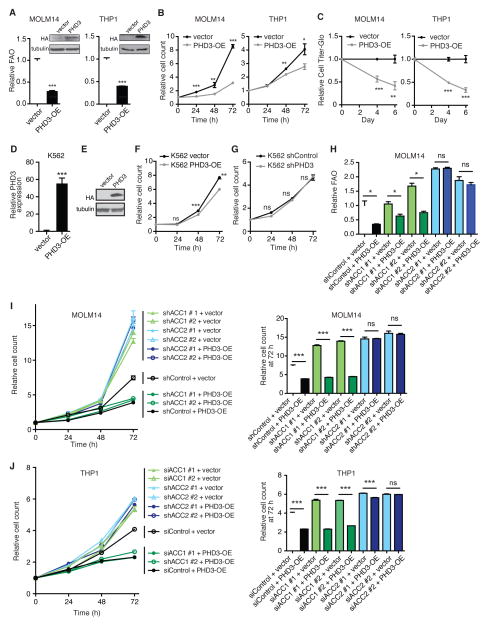
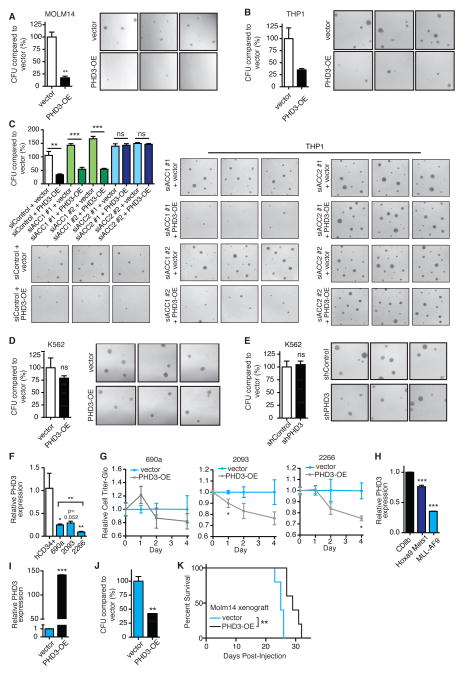
While much research has examined the use of glucose and glutamine by tumor cells, many cancers instead prefer to metabolize fats. Despite the pervasiveness of this phenotype, knowledge of pathways that drive fatty acid oxidation (FAO) in cancer is limited. Prolyl hydroxylase domain proteins hydroxylate substrate proline residues and have been linked to fuel switching. Here, we reveal that PHD3 rapidly triggers repression of FAO in response to nutrient abundance via hydroxylation of acetyl-coA carboxylase 2 (ACC2). We find that PHD3 expression is strongly decreased in subsets of cancer including acute myeloid leukemia (AML) and is linked to a reliance on fat catabolism regardless of external nutrient cues. Overexpressing PHD3 limits FAO via regulation of ACC2 and consequently impedes leukemia cell proliferation. Thus, loss of PHD3 enables greater utilization of fatty acids but may also serve as a metabolic and therapeutic liability by indicating cancer cell susceptibility to FAO inhibition.
Copyright © 2016 Elsevier Inc. All rights reserved.
Figures
References
-
- Almarza-Ortega DB. Human Acetyl-CoA Carboxylase 2. Molecular Cloning, Characterization, Chromosomal Mapping, and Evidence for Two Isoforms. Journal of Biological Chemistry. 1997;272:10669–10677. - PubMed
-
- Andersson A, Ritz C, Lindgren D, Edén P, Lassen C, Heldrup J, Olofsson T, Råde J, Fontes M, Porwit-MacDonald A, et al. Microarray-based classification of a consecutive series of 121 childhood acute leukemias: prediction of leukemic and genetic subtype as well as of minimal residual disease status. Leukemia. 2007;21:1198–1203. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
