

Recurrent De Novo and Biallelic Variation of ATAD3A, Encoding a Mitochondrial Membrane Protein, Results in Distinct Neurological Syndromes
- PMID: 27640307
- PMCID: PMC5065660
- DOI: 10.1016/j.ajhg.2016.08.007
Recurrent De Novo and Biallelic Variation of ATAD3A, Encoding a Mitochondrial Membrane Protein, Results in Distinct Neurological Syndromes
Abstract
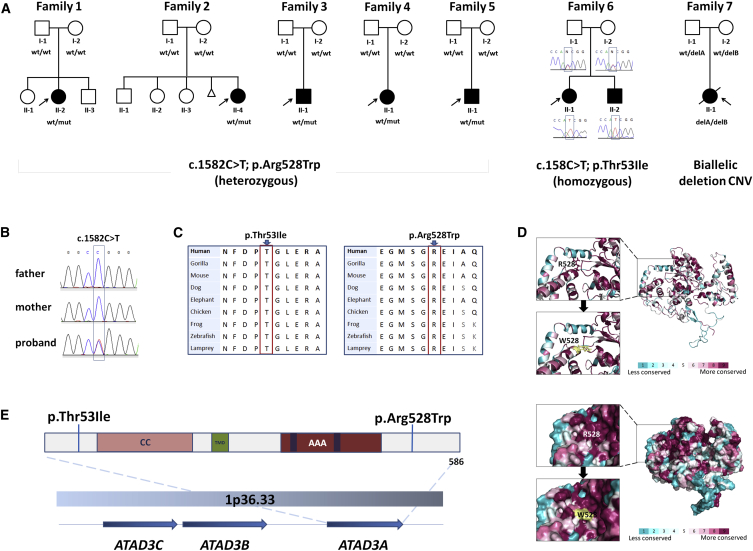
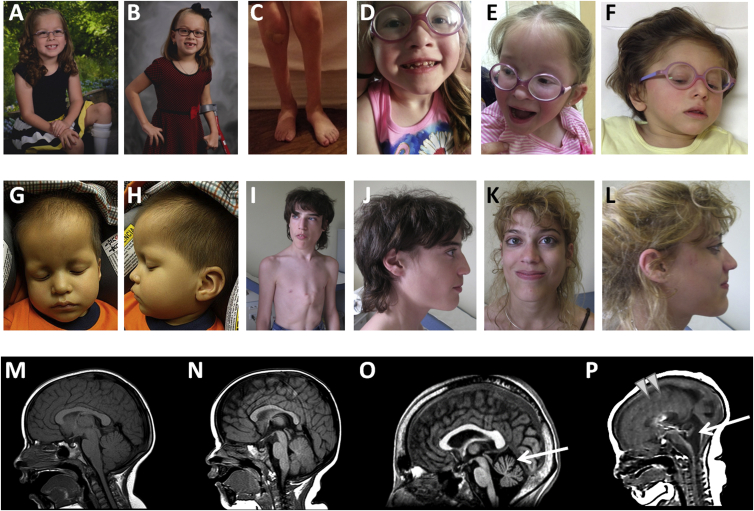
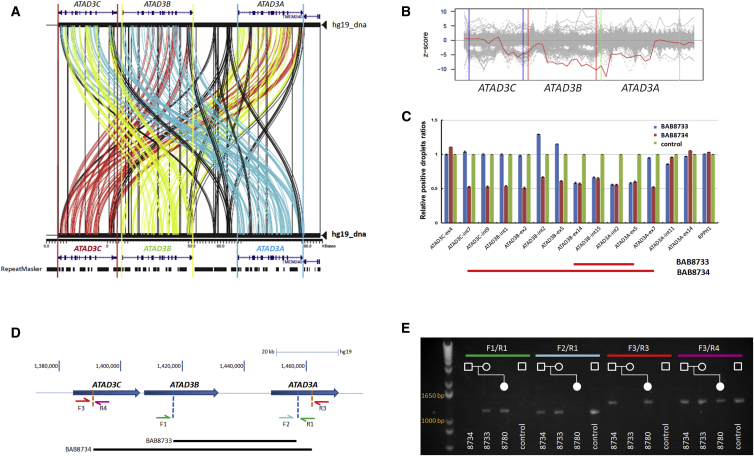
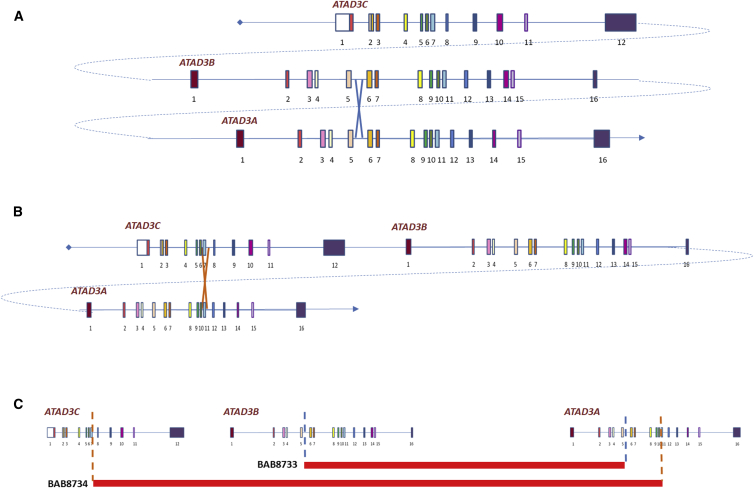
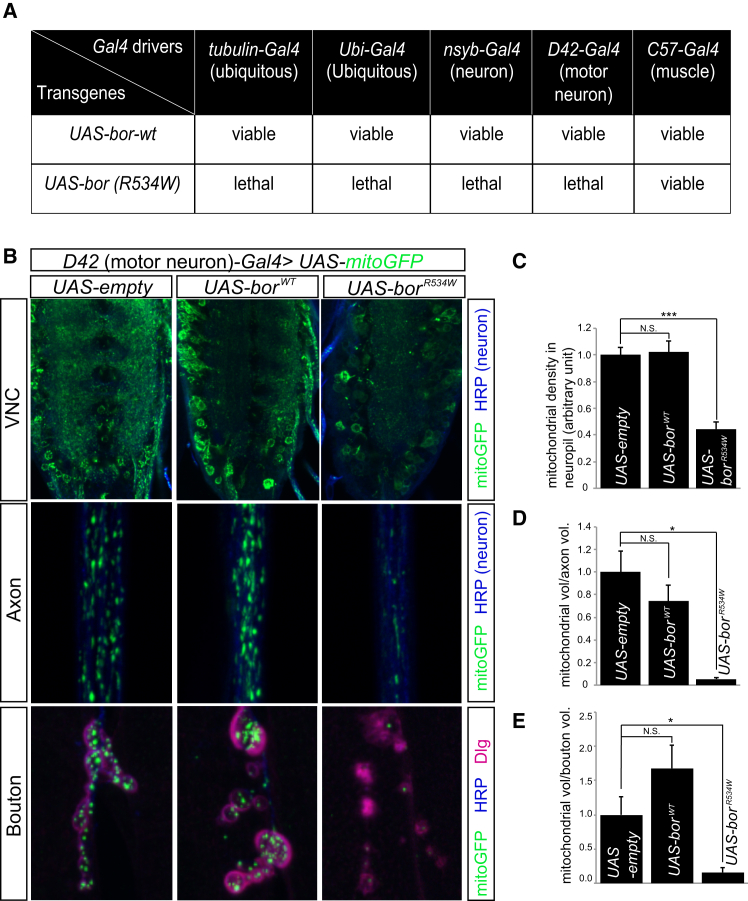
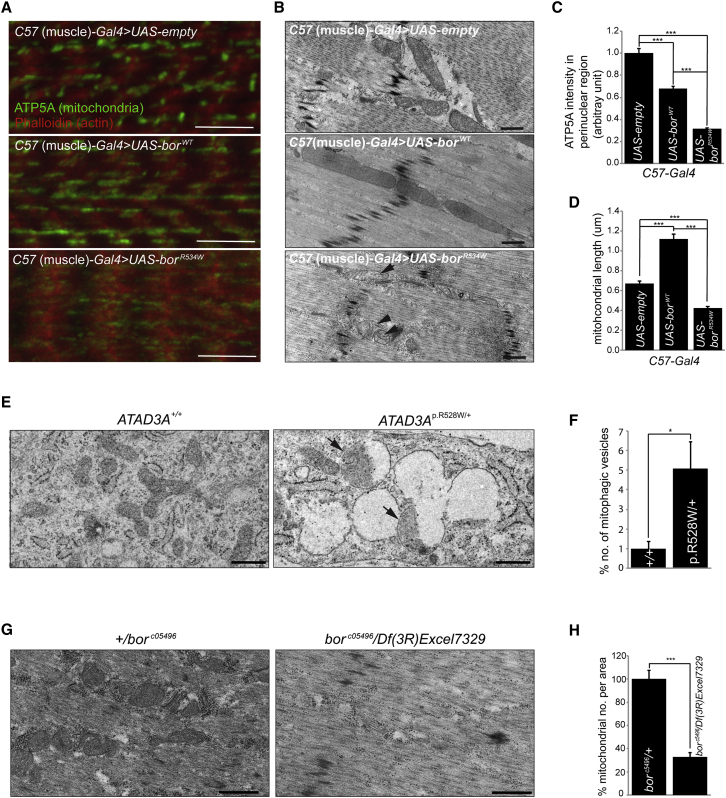
ATPase family AAA-domain containing protein 3A (ATAD3A) is a nuclear-encoded mitochondrial membrane protein implicated in mitochondrial dynamics, nucleoid organization, protein translation, cell growth, and cholesterol metabolism. We identified a recurrent de novo ATAD3A c.1582C>T (p.Arg528Trp) variant by whole-exome sequencing (WES) in five unrelated individuals with a core phenotype of global developmental delay, hypotonia, optic atrophy, axonal neuropathy, and hypertrophic cardiomyopathy. We also describe two families with biallelic variants in ATAD3A, including a homozygous variant in two siblings, and biallelic ATAD3A deletions mediated by nonallelic homologous recombination (NAHR) between ATAD3A and gene family members ATAD3B and ATAD3C. Tissue-specific overexpression of borR534W, the Drosophila mutation homologous to the human c.1582C>T (p.Arg528Trp) variant, resulted in a dramatic decrease in mitochondrial content, aberrant mitochondrial morphology, and increased autophagy. Homozygous null bor larvae showed a significant decrease of mitochondria, while overexpression of borWT resulted in larger, elongated mitochondria. Finally, fibroblasts of an affected individual exhibited increased mitophagy. We conclude that the p.Arg528Trp variant functions through a dominant-negative mechanism that results in small mitochondria that trigger mitophagy, resulting in a reduction in mitochondrial content. ATAD3A variation represents an additional link between mitochondrial dynamics and recognizable neurological syndromes, as seen with MFN2, OPA1, DNM1L, and STAT2 mutations.
Keywords: ATAD3A; CNV; cardiomyopathy; de novo variant; dominant negative; mitochondrial dynamics; neuropathy; optic atrophy; whole-exome sequencing.
Copyright © 2016 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Pareyson D., Saveri P., Sagnelli A., Piscosquito G. Mitochondrial dynamics and inherited peripheral nerve diseases. Neurosci. Lett. 2015;596:66–77. - PubMed
-
- Züchner S., Mersiyanova I.V., Muglia M., Bissar-Tadmouri N., Rochelle J., Dadali E.L., Zappia M., Nelis E., Patitucci A., Senderek J. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 2004;36:449–451. - PubMed
-
- Züchner S., De Jonghe P., Jordanova A., Claeys K.G., Guergueltcheva V., Cherninkova S., Hamilton S.R., Van Stavern G., Krajewski K.M., Stajich J. Axonal neuropathy with optic atrophy is caused by mutations in mitofusin 2. Ann. Neurol. 2006;59:276–281. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
