

A Biobank of Breast Cancer Explants with Preserved Intra-tumor Heterogeneity to Screen Anticancer Compounds
- PMID: 27641504
- PMCID: PMC5037319
- DOI: 10.1016/j.cell.2016.08.041
A Biobank of Breast Cancer Explants with Preserved Intra-tumor Heterogeneity to Screen Anticancer Compounds
Abstract
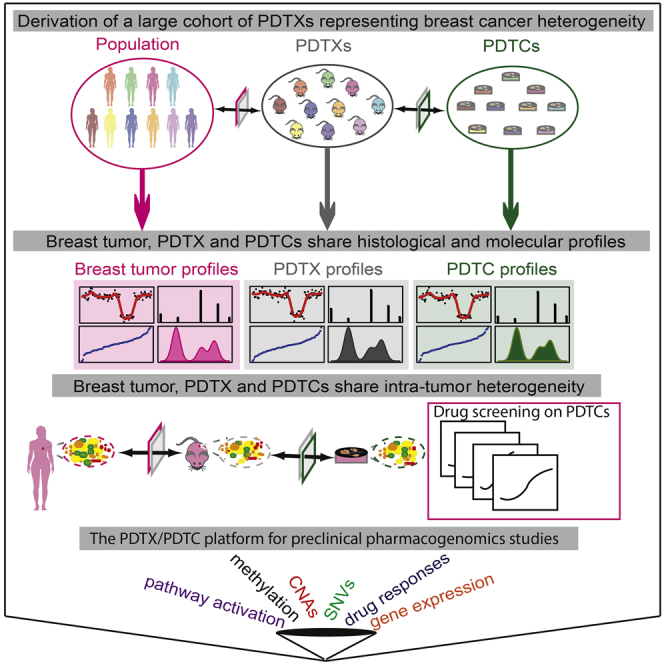
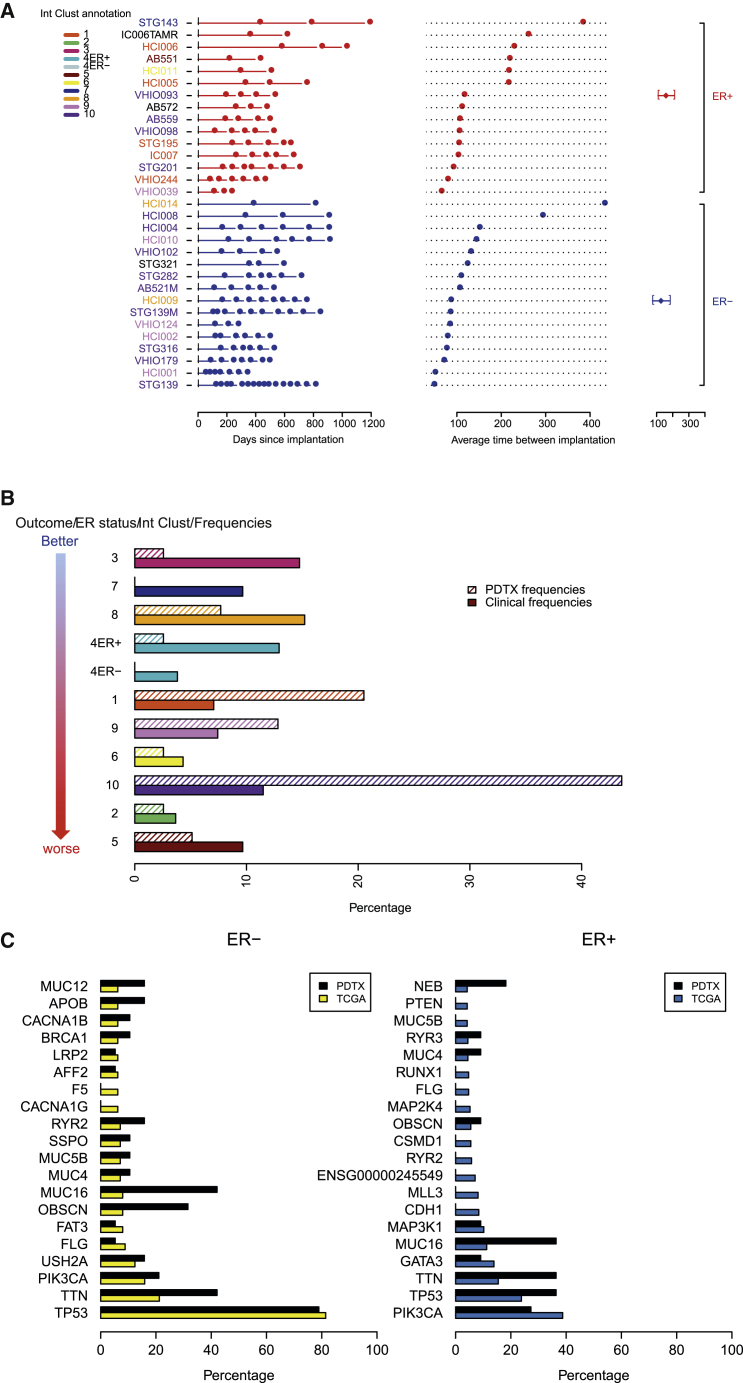
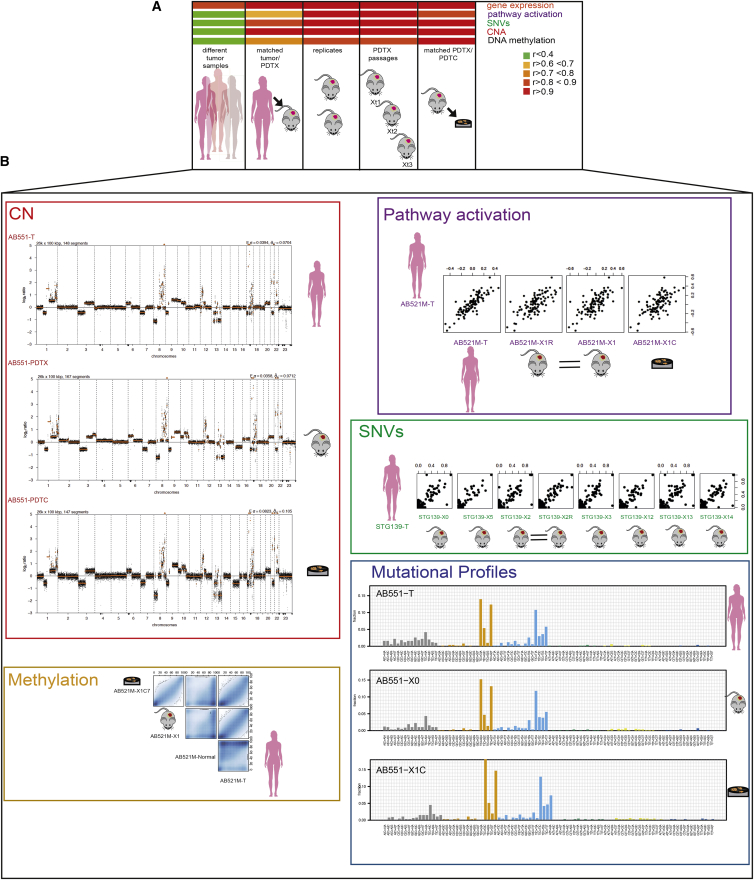
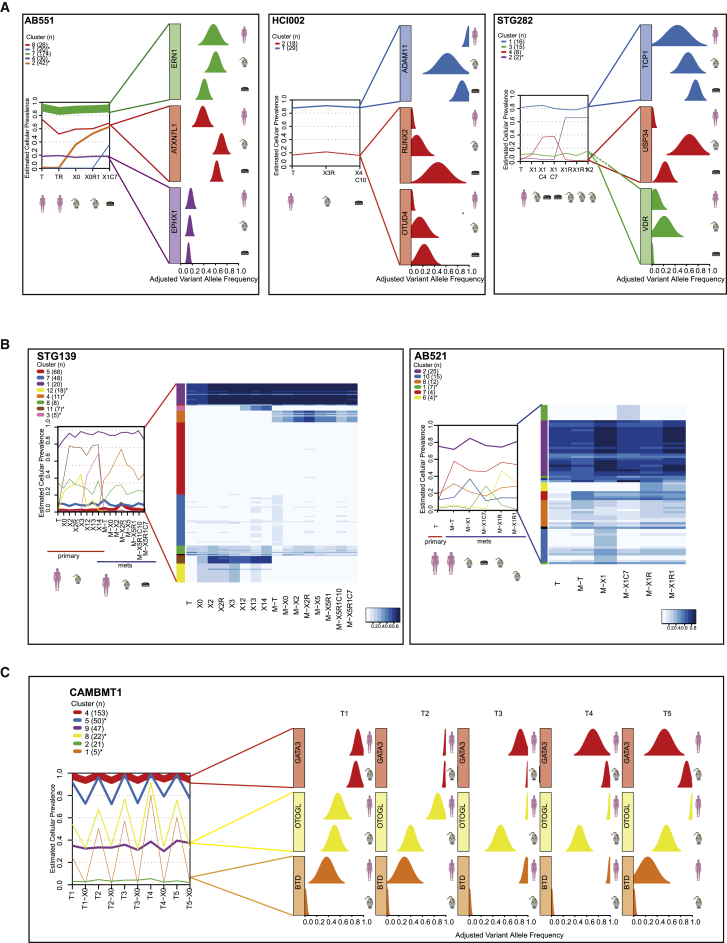
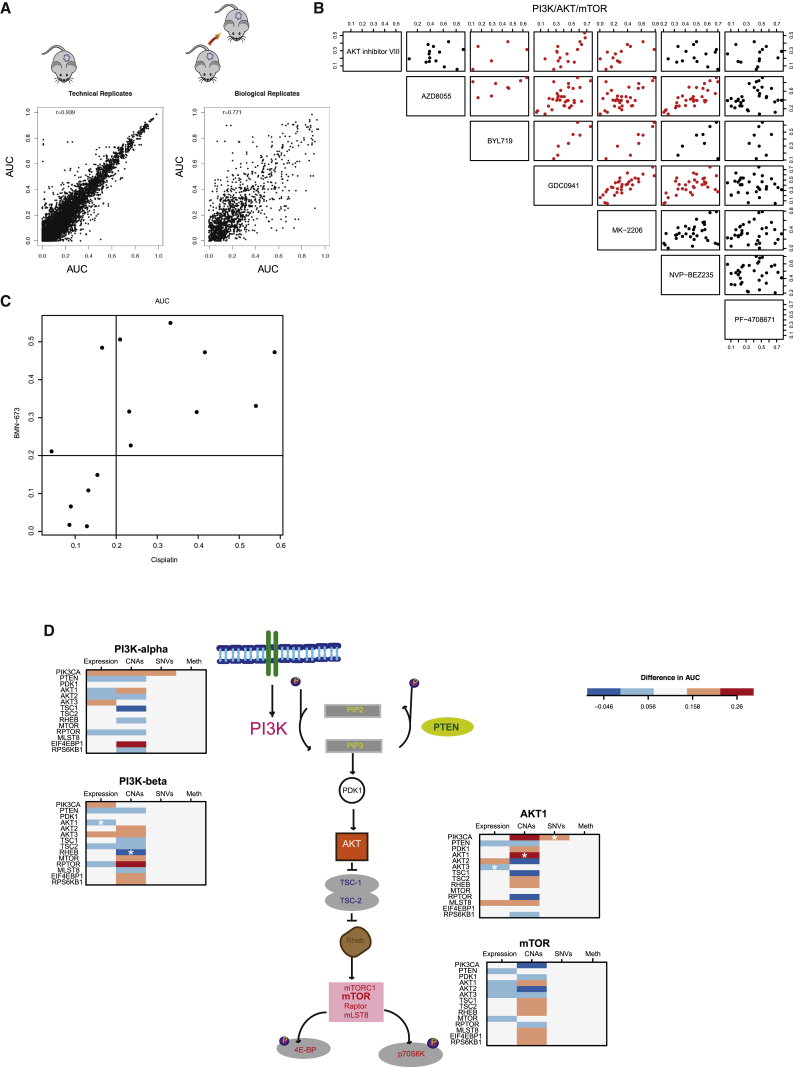
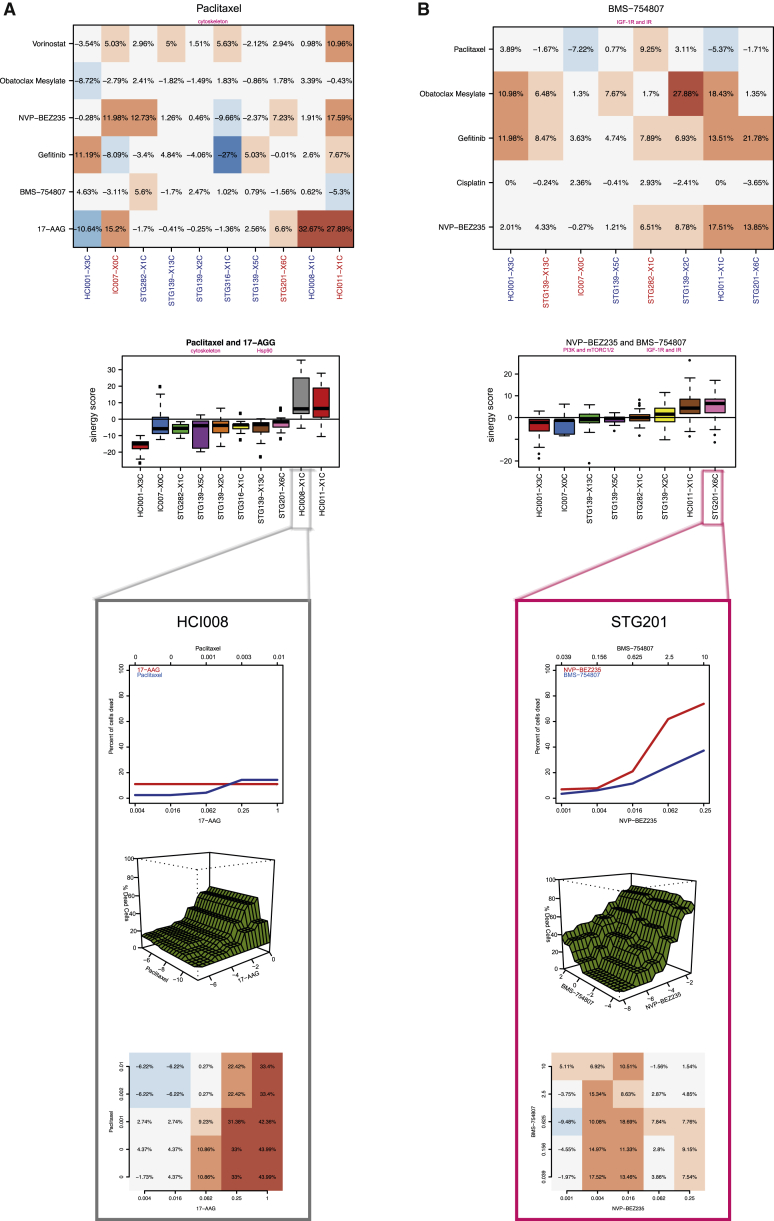
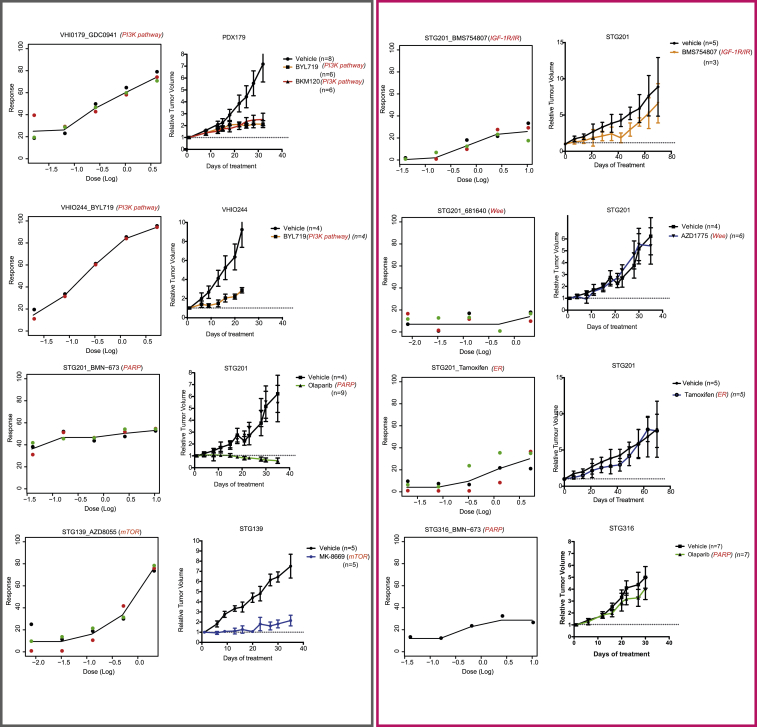
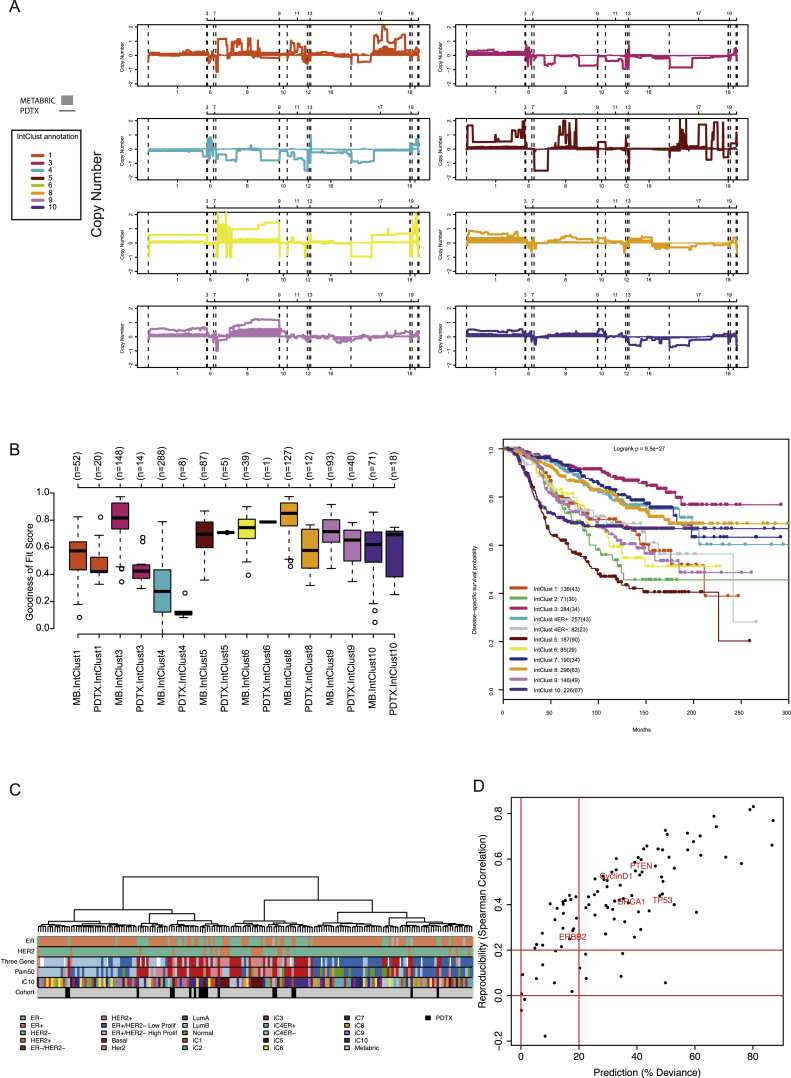
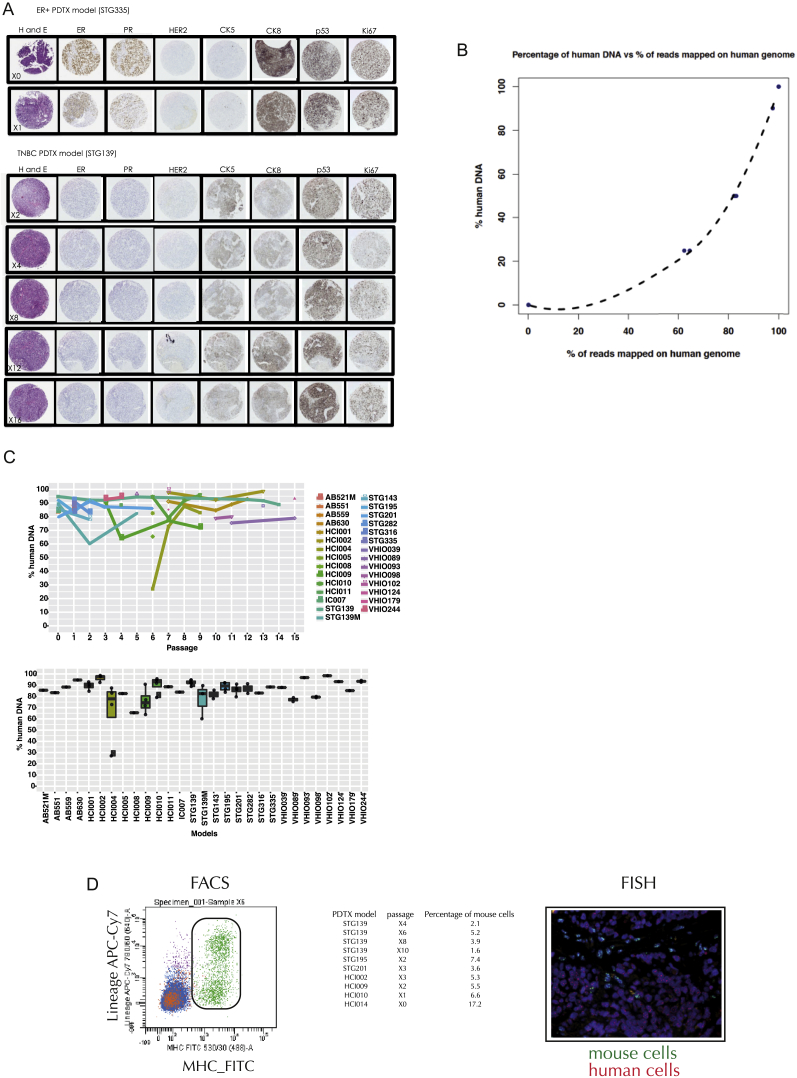
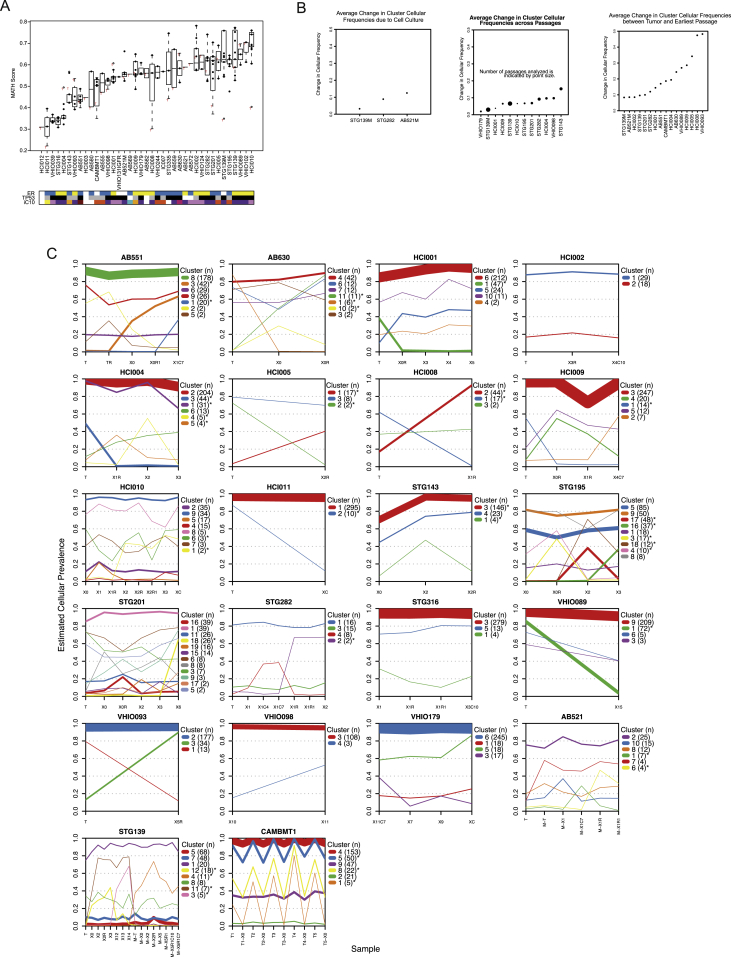
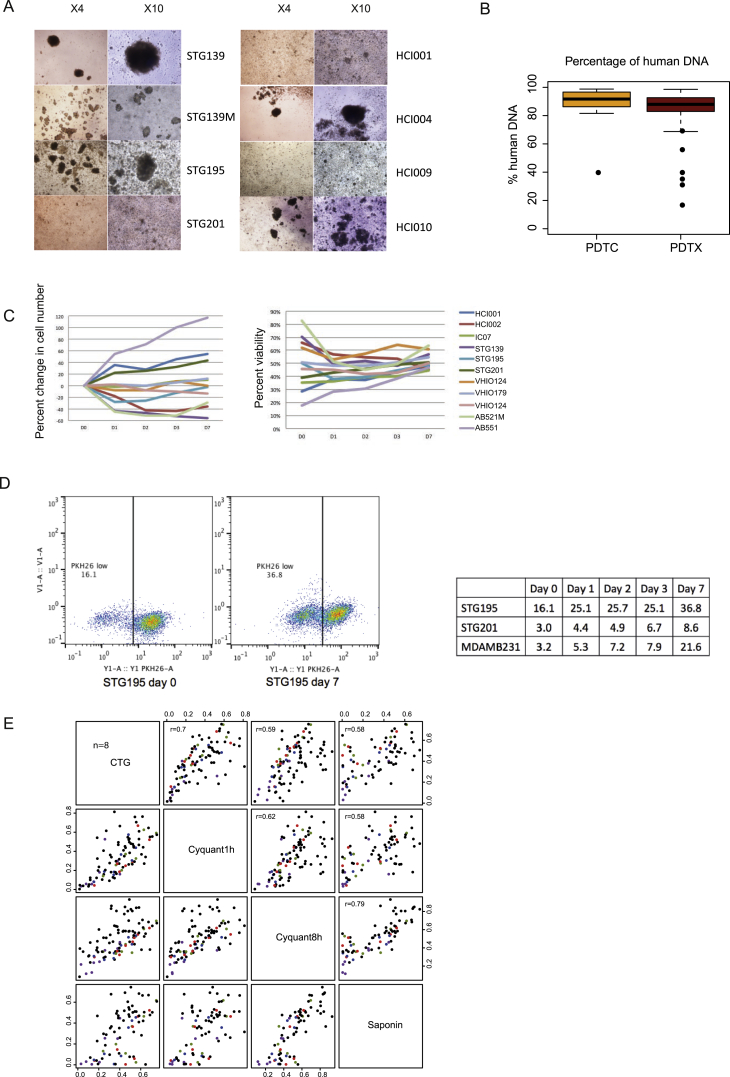
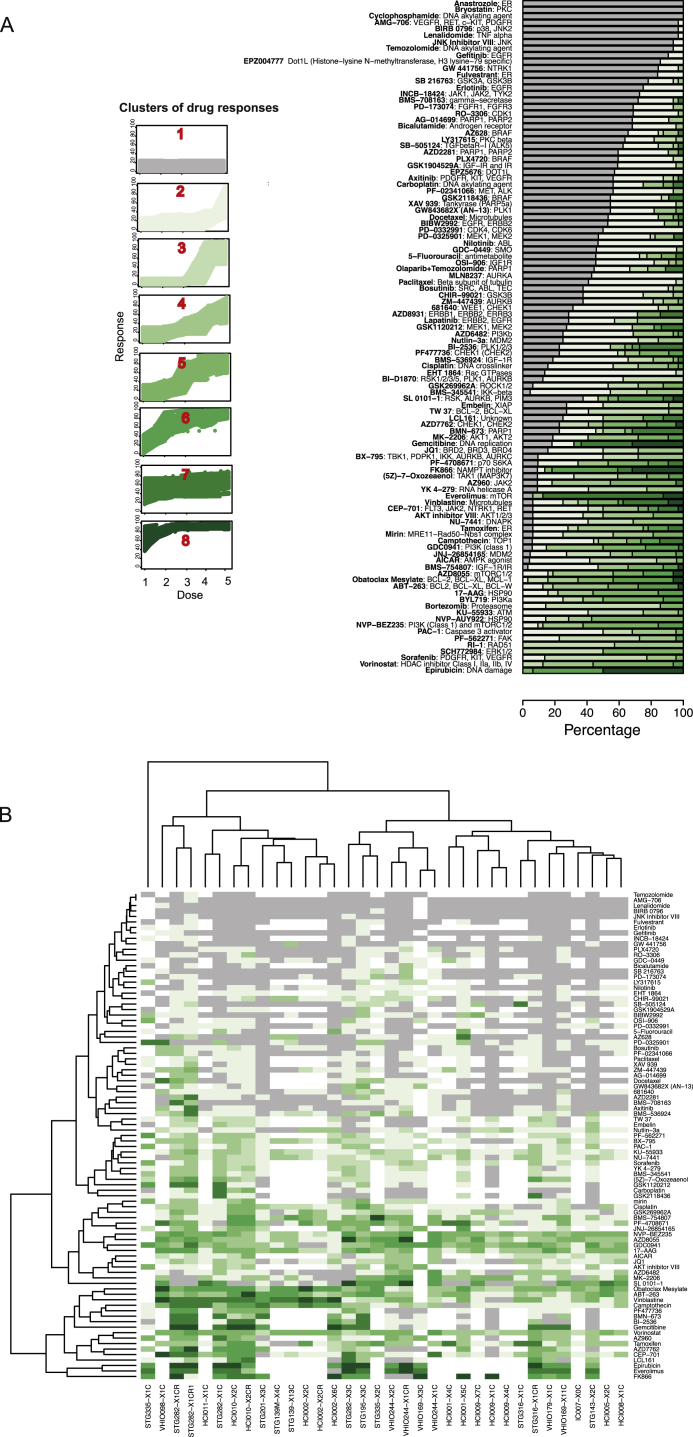
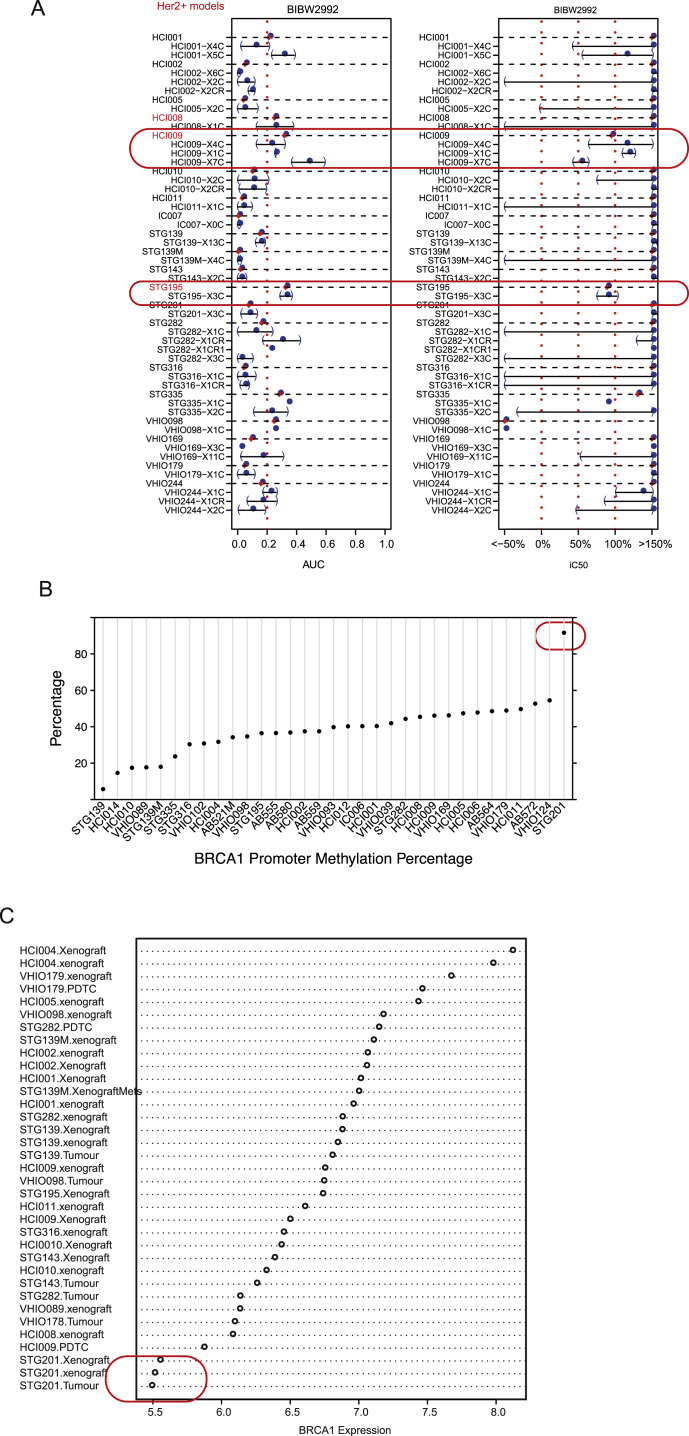
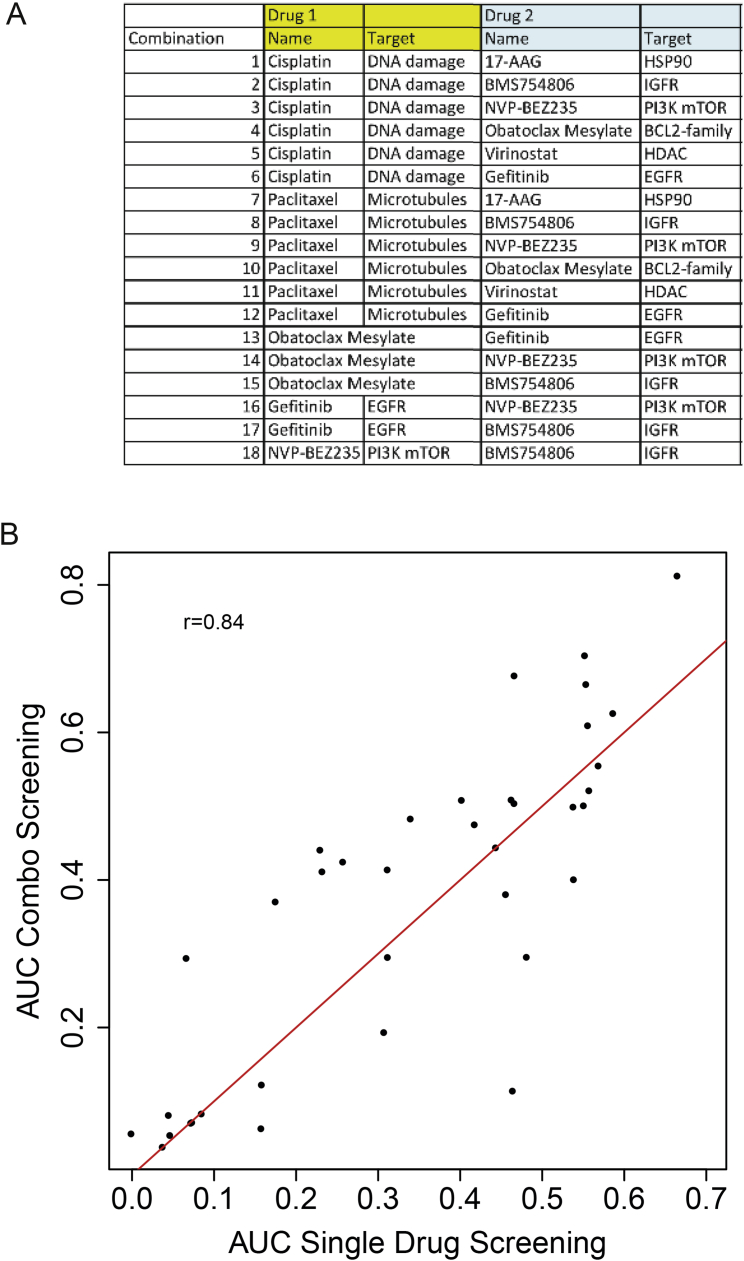
The inter- and intra-tumor heterogeneity of breast cancer needs to be adequately captured in pre-clinical models. We have created a large collection of breast cancer patient-derived tumor xenografts (PDTXs), in which the morphological and molecular characteristics of the originating tumor are preserved through passaging in the mouse. An integrated platform combining in vivo maintenance of these PDTXs along with short-term cultures of PDTX-derived tumor cells (PDTCs) was optimized. Remarkably, the intra-tumor genomic clonal architecture present in the originating breast cancers was mostly preserved upon serial passaging in xenografts and in short-term cultured PDTCs. We assessed drug responses in PDTCs on a high-throughput platform and validated several ex vivo responses in vivo. The biobank represents a powerful resource for pre-clinical breast cancer pharmacogenomic studies (http://caldaslab.cruk.cam.ac.uk/bcape), including identification of biomarkers of response or resistance.
Copyright © 2016 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Alexandrov L.B., Nik-Zainal S., Wedge D.C., Aparicio S.A., Behjati S., Biankin A.V., Bignell G.R., Bolli N., Borg A., Børresen-Dale A.L., Australian Pancreatic Cancer Genome Initiative. ICGC Breast Cancer Consortium. ICGC MMML-Seq Consortium. ICGC PedBrain Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. - PMC - PubMed
-
- Aparicio S., Caldas C. The implications of clonal genome evolution for cancer medicine. N. Engl. J. Med. 2013;368:842–851. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
