

In vivo disruption of latent HSV by designer endonuclease therapy
- PMID: 27642635
- PMCID: PMC5026126
- DOI: 10.1172/jci.insight.88468
In vivo disruption of latent HSV by designer endonuclease therapy
Abstract
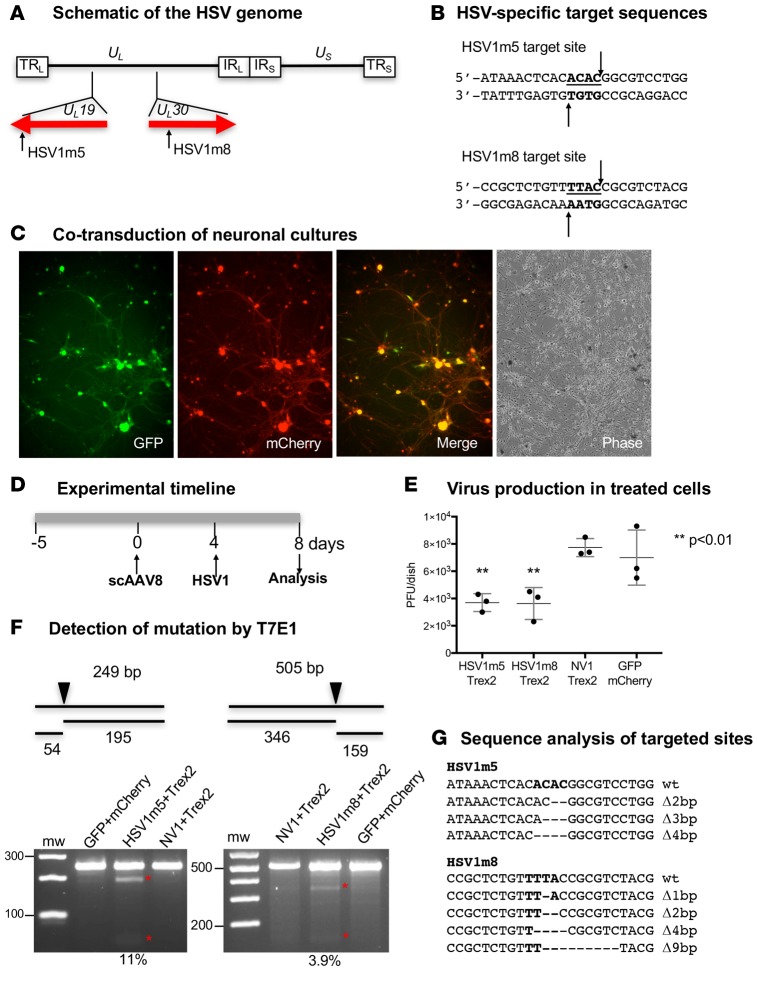
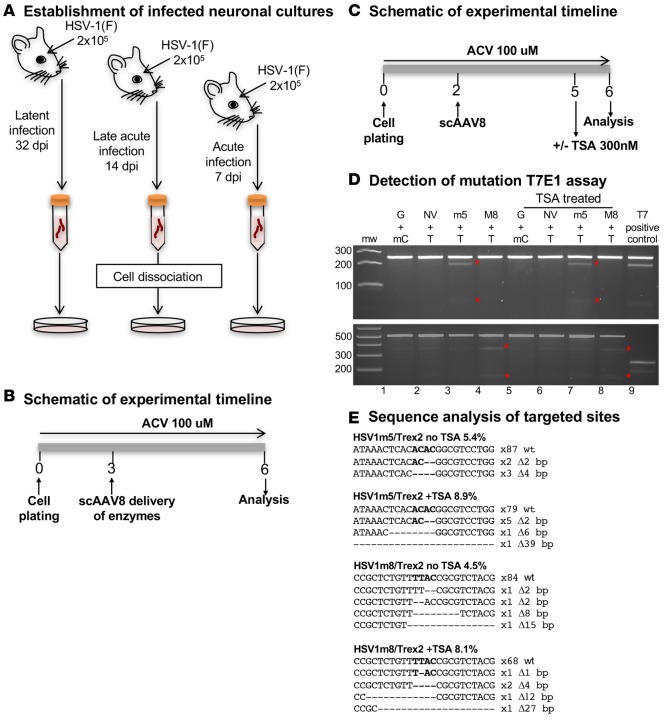
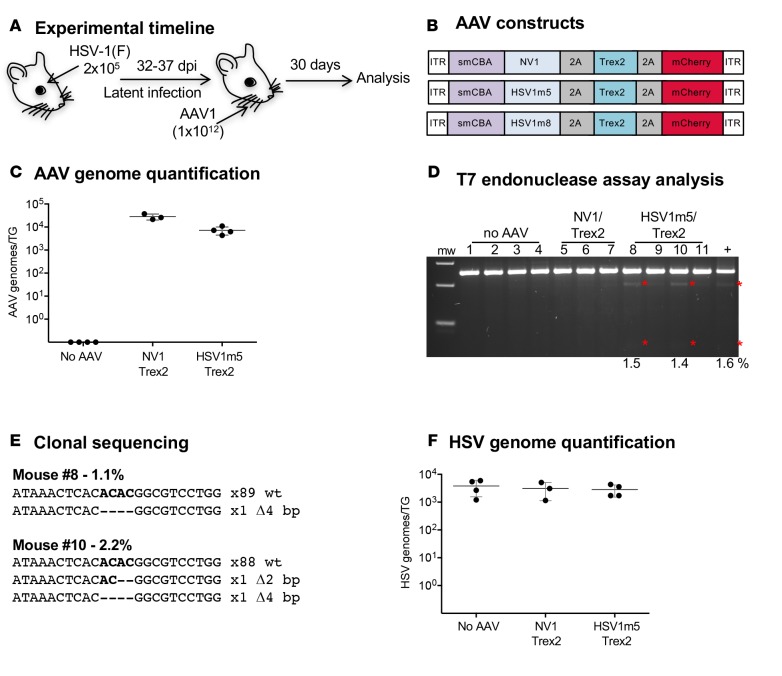
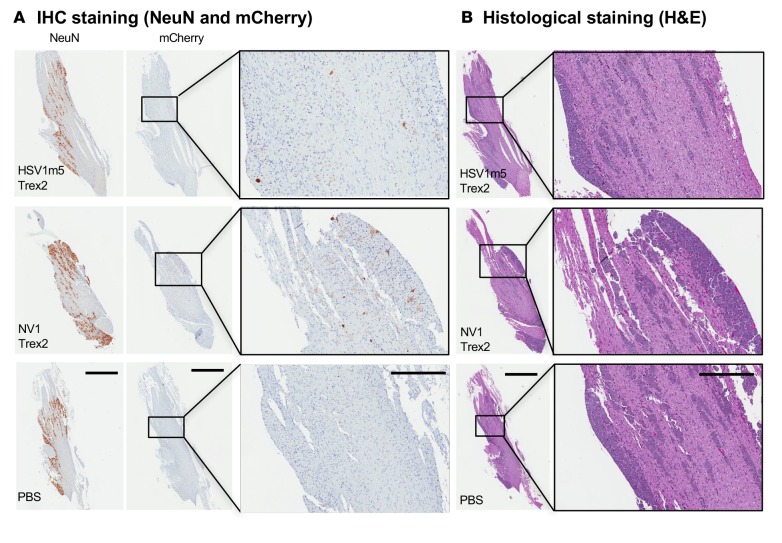
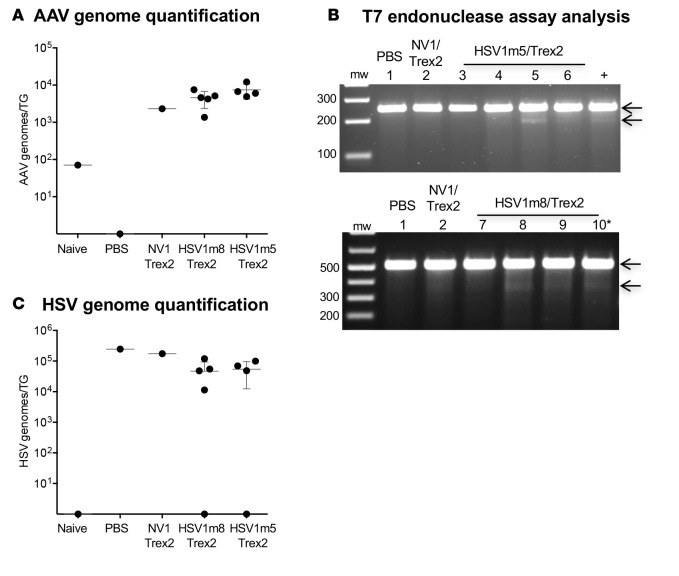
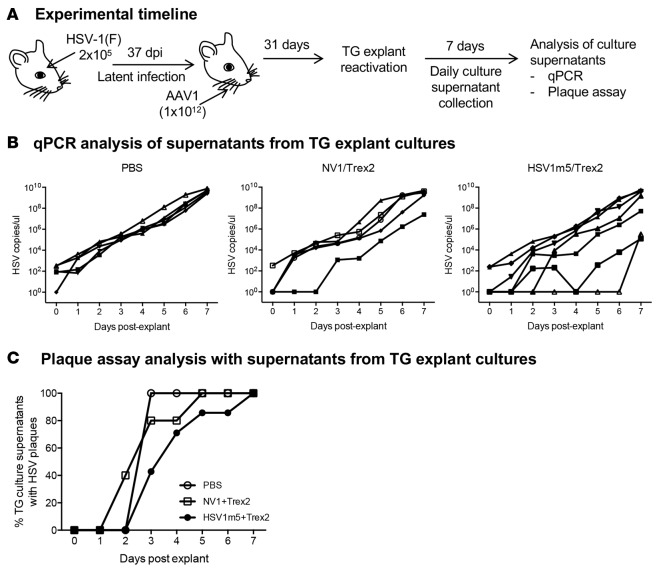
A large portion of the global population carries latent herpes simplex virus (HSV), which can periodically reactivate, resulting in asymptomatic shedding or formation of ulcerative lesions. Current anti-HSV drugs do not eliminate latent virus from sensory neurons where HSV resides, and therefore do not eliminate the risk of transmission or recurrent disease. Here, we report the ability of HSV-specific endonucleases to induce mutations of essential HSV genes both in cultured neurons and in latently infected mice. In neurons, viral genomes are susceptible to endonuclease-mediated mutagenesis, regardless of the time of treatment after HSV infection, suggesting that both HSV lytic and latent forms can be targeted. Mutagenesis frequency after endonuclease exposure can be increased nearly 2-fold by treatment with a histone deacetylase (HDAC) inhibitor. Using a mouse model of latent HSV infection, we demonstrate that a targeted endonuclease can be delivered to viral latency sites via an adeno-associated virus (AAV) vector, where it is able to induce mutation of latent HSV genomes. These data provide the first proof-of-principle to our knowledge for the use of a targeted endonuclease as an antiviral agent to treat an established latent viral infection in vivo.
Conflict of interest statement
The authors have declared that no conflict of interest exists.
Figures

References
-
- Roizman B, Knipe DM, Whitley RJ. 2007. Herpes Simplex Viruses. In: Knipe DM, Howley PM, eds. Fields Virology. Wolters Kluwer Health/Lippincott Williams & Wilkins; 2007:2502–2599.
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
