

Hepatocellular carcinoma redirects to ketolysis for progression under nutrition deprivation stress
- PMID: 27644987
- PMCID: PMC5113304
- DOI: 10.1038/cr.2016.109
Hepatocellular carcinoma redirects to ketolysis for progression under nutrition deprivation stress
Abstract
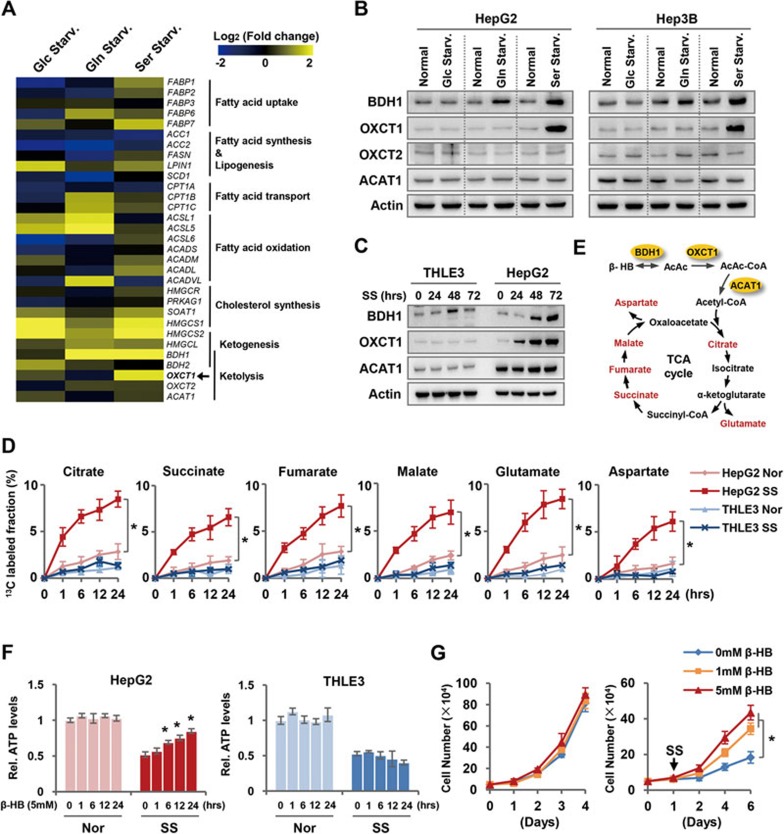
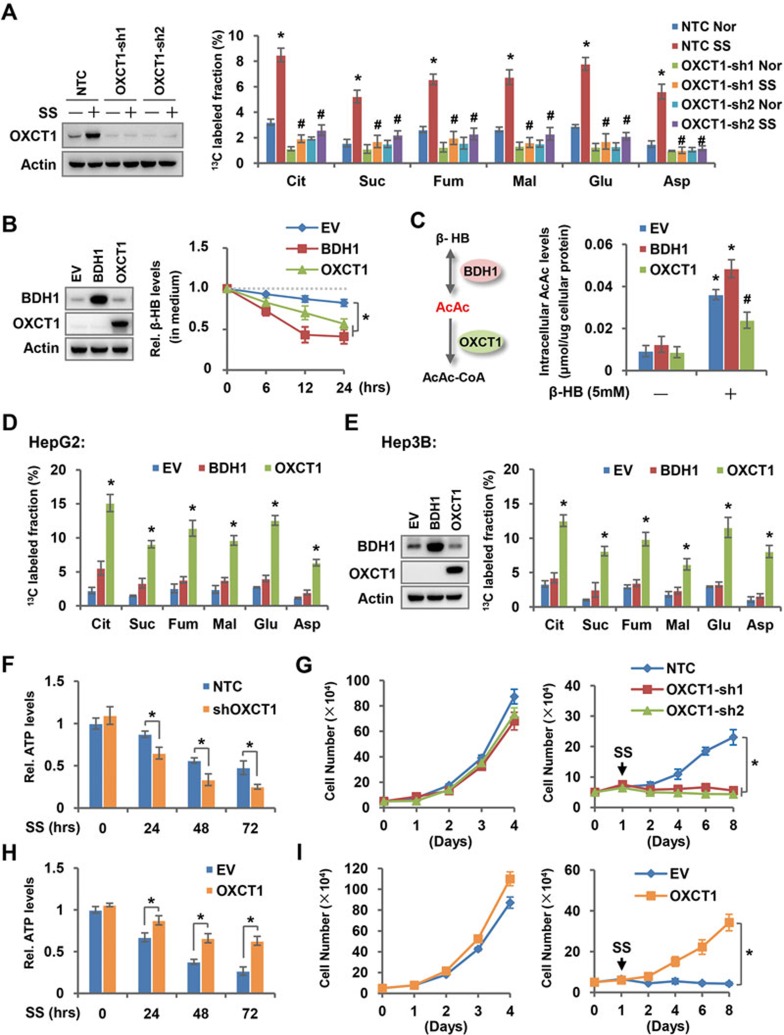
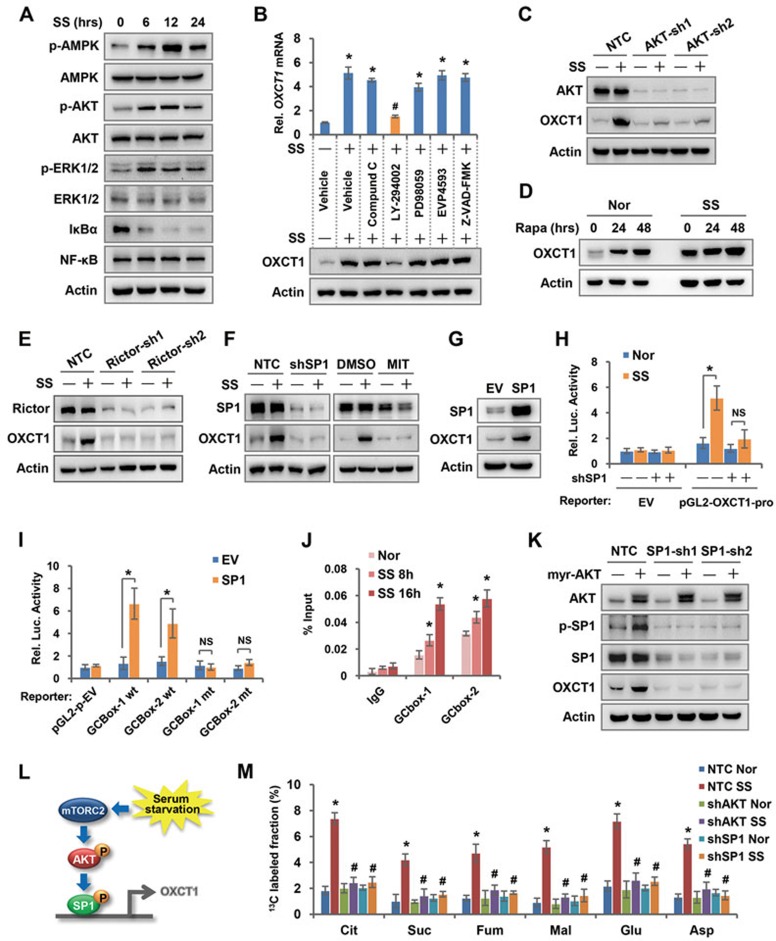
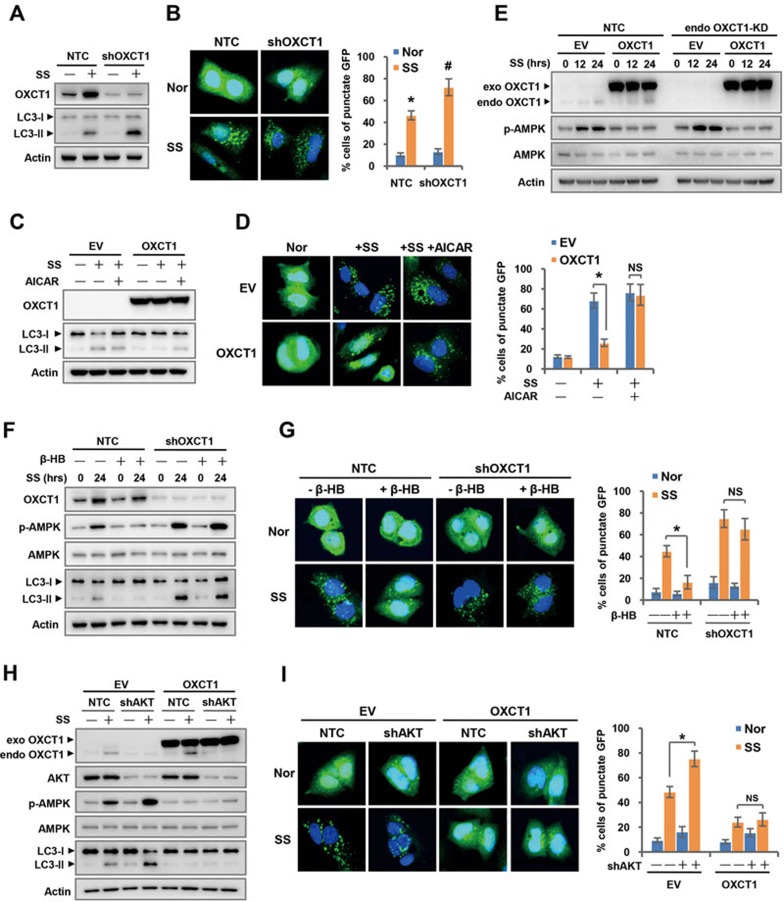
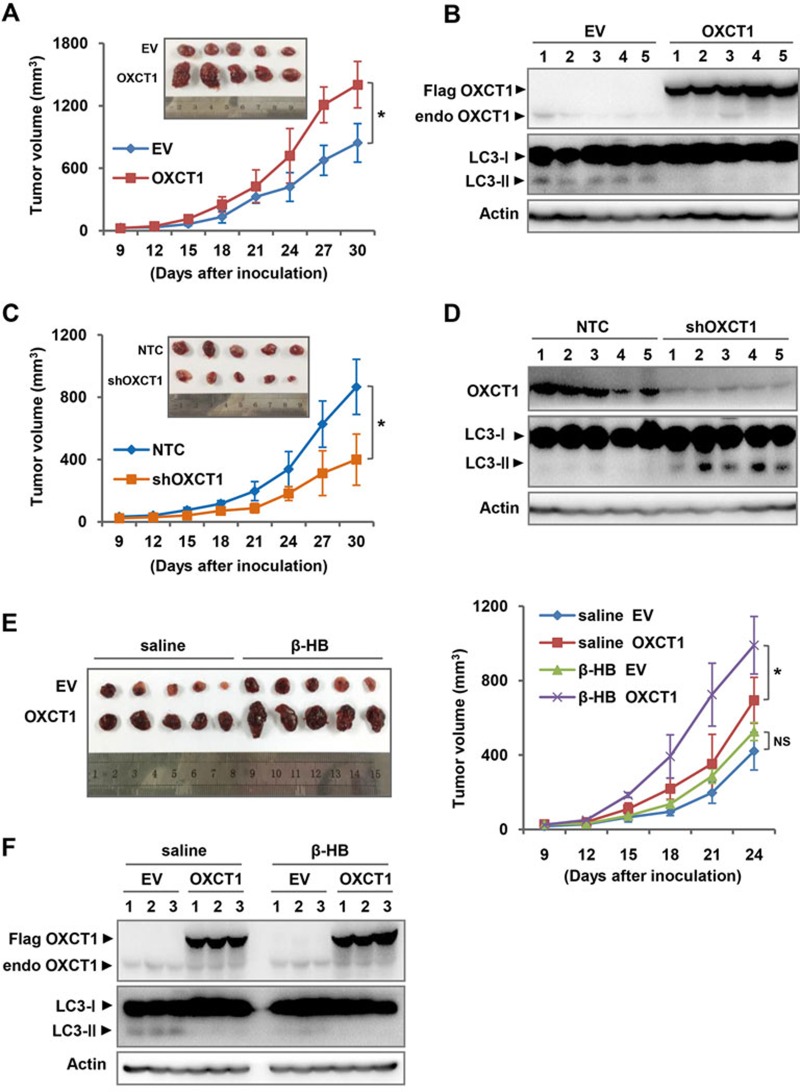
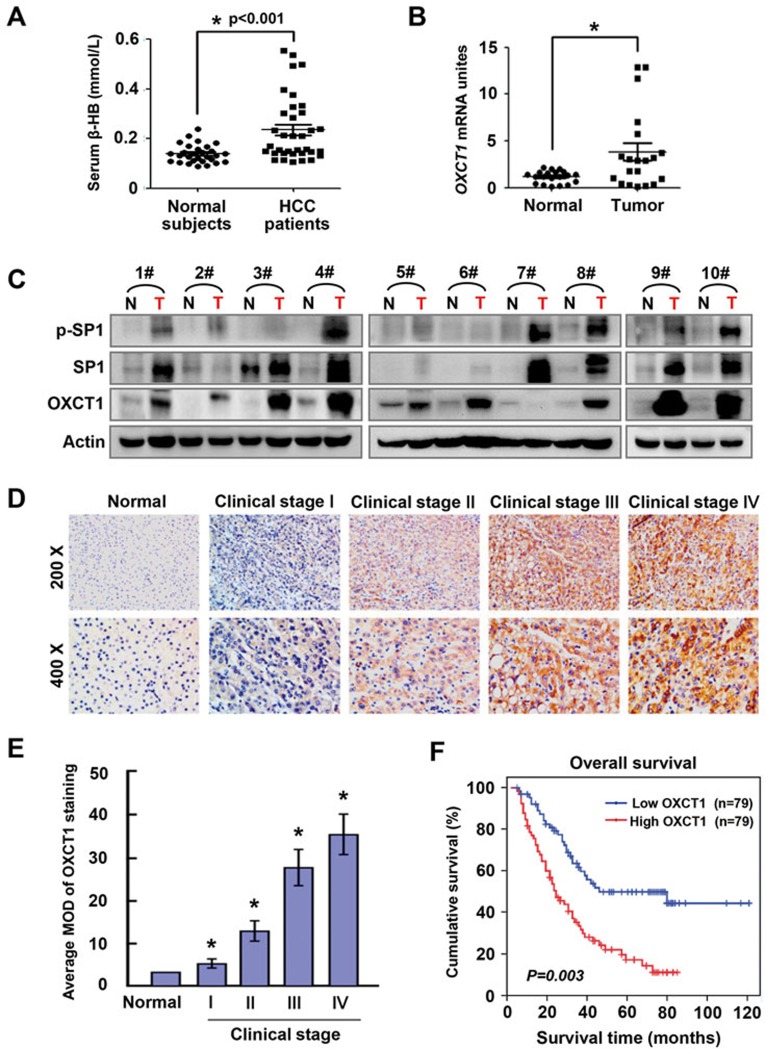
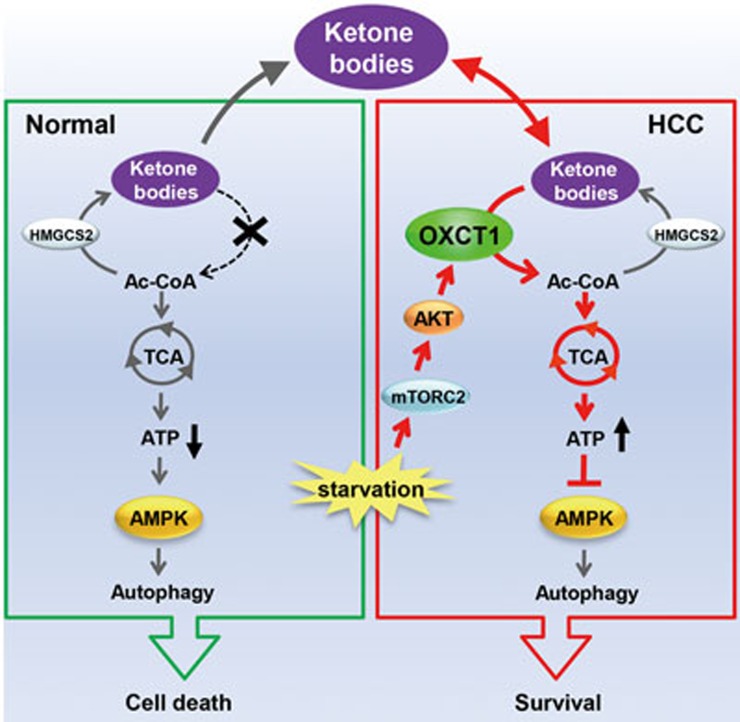
Cancer cells are known for their capacity to rewire metabolic pathways to support survival and proliferation under various stress conditions. Ketone bodies, though produced in the liver, are not consumed in normal adult liver cells. We find here that ketone catabolism or ketolysis is re-activated in hepatocellular carcinoma (HCC) cells under nutrition deprivation conditions. Mechanistically, 3-oxoacid CoA-transferase 1 (OXCT1), a rate-limiting ketolytic enzyme whose expression is suppressed in normal adult liver tissues, is re-induced by serum starvation-triggered mTORC2-AKT-SP1 signaling in HCC cells. Moreover, we observe that enhanced ketolysis in HCC is critical for repression of AMPK activation and protects HCC cells from excessive autophagy, thereby enhancing tumor growth. Importantly, analysis of clinical HCC samples reveals that increased OXCT1 expression predicts higher patient mortality. Taken together, we uncover here a novel metabolic adaptation by which nutrition-deprived HCC cells employ ketone bodies for energy supply and cancer progression.
Figures
Comment in
-
Aberrant ketolysis fuels hepatocellular cancer progression.Cell Res. 2016 Oct;26(10):1077-1078. doi: 10.1038/cr.2016.110. Epub 2016 Sep 20. Cell Res. 2016. PMID: 27644988 Free PMC article.
Similar articles
-
OXCT1 succinylation and activation by SUCLA2 promotes ketolysis and liver tumor growth.Mol Cell. 2025 Feb 20;85(4):843-856.e6. doi: 10.1016/j.molcel.2024.12.025. Epub 2025 Jan 24. Mol Cell. 2025. PMID: 39862868
-
Targeting OXCT1-mediated ketone metabolism reprograms macrophages to promote antitumor immunity via CD8+ T cells in hepatocellular carcinoma.J Hepatol. 2024 Oct;81(4):690-703. doi: 10.1016/j.jhep.2024.05.007. Epub 2024 May 15. J Hepatol. 2024. PMID: 38759889
-
AKT activation was not essential for hepatocellular carcinoma cell survival under glucose deprivation.Anticancer Drugs. 2017 Apr;28(4):427-435. doi: 10.1097/CAD.0000000000000475. Anticancer Drugs. 2017. PMID: 28085697
-
The role of OXCT1 in the pathogenesis of cancer as a rate-limiting enzyme of ketone body metabolism.Life Sci. 2017 Aug 15;183:110-115. doi: 10.1016/j.lfs.2017.07.003. Epub 2017 Jul 4. Life Sci. 2017. PMID: 28684065 Review.
-
Succinyl-CoA:3-oxoacid coenzyme A transferase (SCOT) deficiency: A rare and potentially fatal metabolic disease.Biochimie. 2021 Apr;183:55-62. doi: 10.1016/j.biochi.2021.02.003. Epub 2021 Feb 14. Biochimie. 2021. PMID: 33596448
Cited by
-
The roles of metabolic profiles and intracellular signaling pathways of tumor microenvironment cells in angiogenesis of solid tumors.Cell Commun Signal. 2022 Nov 23;20(1):186. doi: 10.1186/s12964-022-00951-y. Cell Commun Signal. 2022. PMID: 36419156 Free PMC article. Review.
-
Critical Investigation of the Usability of Hepatoma Cell Lines HepG2 and Huh7 as Models for the Metabolic Representation of Resectable Hepatocellular Carcinoma.Cancers (Basel). 2022 Aug 30;14(17):4227. doi: 10.3390/cancers14174227. Cancers (Basel). 2022. PMID: 36077764 Free PMC article.
-
Ketogenic Diets and Hepatocellular Carcinoma.Front Oncol. 2022 May 4;12:879205. doi: 10.3389/fonc.2022.879205. eCollection 2022. Front Oncol. 2022. PMID: 35600387 Free PMC article. Review.
-
Influence of the Tumor Microenvironment on Cancer Cells Metabolic Reprogramming.Front Oncol. 2018 Apr 19;8:117. doi: 10.3389/fonc.2018.00117. eCollection 2018. Front Oncol. 2018. PMID: 29725585 Free PMC article. Review.
-
Unrestricted Ketogenic Diet Feeding Enhances Epithelial Ovarian Cancer Growth In Vivo.Nutrients. 2023 Jun 13;15(12):2730. doi: 10.3390/nu15122730. Nutrients. 2023. PMID: 37375634 Free PMC article.
References
-
- Lyssiotis CA, Cantley LC. Acetate fuels the cancer engine. Cell 2014; 159:1492–1494. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
