

Direct chromosome-length haplotyping by single-cell sequencing
- PMID: 27646535
- PMCID: PMC5088598
- DOI: 10.1101/gr.209841.116
Direct chromosome-length haplotyping by single-cell sequencing
Abstract
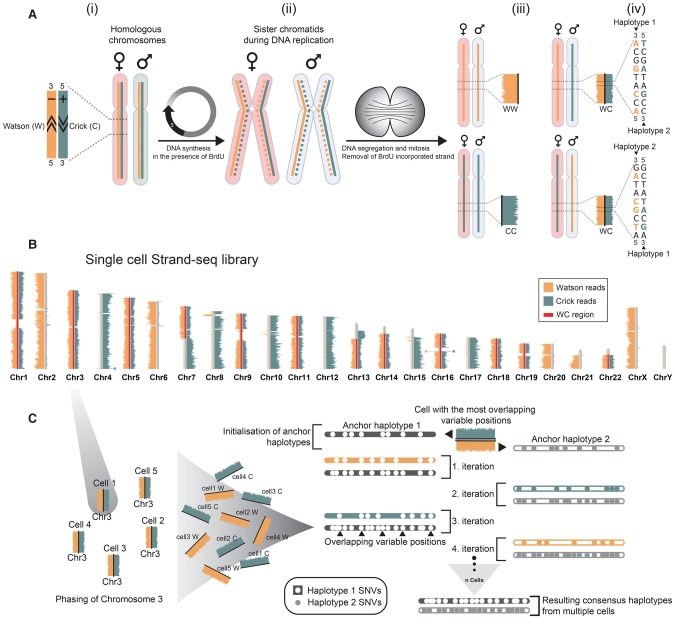
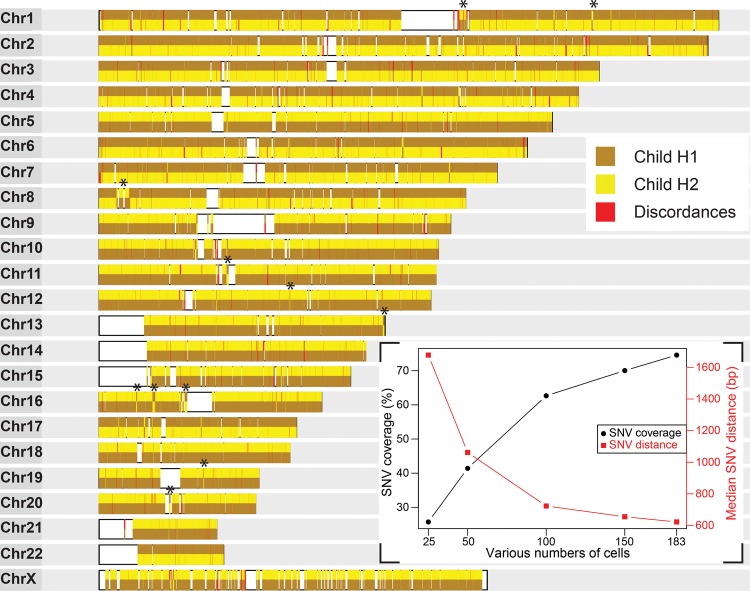
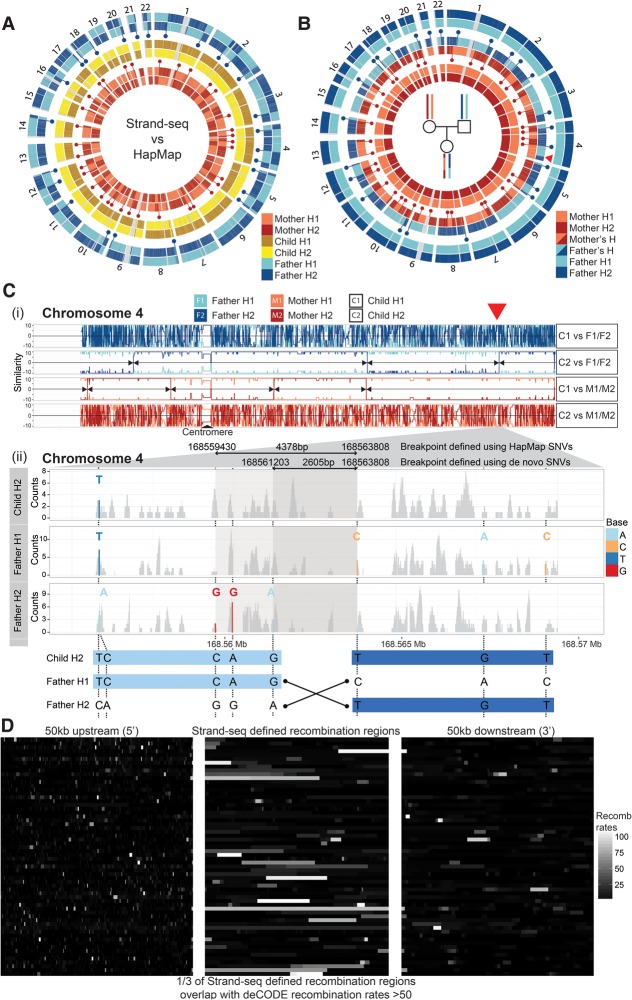
Haplotypes are fundamental to fully characterize the diploid genome of an individual, yet methods to directly chart the unique genetic makeup of each parental chromosome are lacking. Here we introduce single-cell DNA template strand sequencing (Strand-seq) as a novel approach to phasing diploid genomes along the entire length of all chromosomes. We demonstrate this by building a complete haplotype for a HapMap individual (NA12878) at high accuracy (concordance 99.3%), without using generational information or statistical inference. By use of this approach, we mapped all meiotic recombination events in a family trio with high resolution (median range ∼14 kb) and phased larger structural variants like deletions, indels, and balanced rearrangements like inversions. Lastly, the single-cell resolution of Strand-seq allowed us to observe loss of heterozygosity regions in a small number of cells, a significant advantage for studies of heterogeneous cell populations, such as cancer cells. We conclude that Strand-seq is a unique and powerful approach to completely phase individual genomes and map inheritance patterns in families, while preserving haplotype differences between single cells.
© 2016 Porubský et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Bansal V, Tewhey R, Topol EJ, Schork NJ. 2011. The next phase in human genetics. Nat Biotechnol 29: 38–39. - PubMed
-
- Brown PJB, De Pedro MA, Kysela T, Van Der Henst C, Kim J, De Bolle X, Fuqua C, Brun YV. 2012. Completely phased genome sequencing through chromosome sorting. Proc Natl Acad Sci 109: 3190–3190.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources