

NGS-based reverse genetic screen for common embryonic lethal mutations compromising fertility in livestock
- PMID: 27646536
- PMCID: PMC5052051
- DOI: 10.1101/gr.207076.116
NGS-based reverse genetic screen for common embryonic lethal mutations compromising fertility in livestock
Abstract
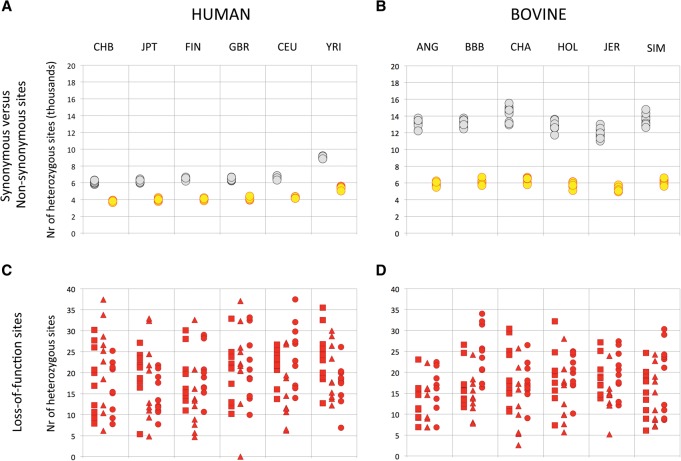
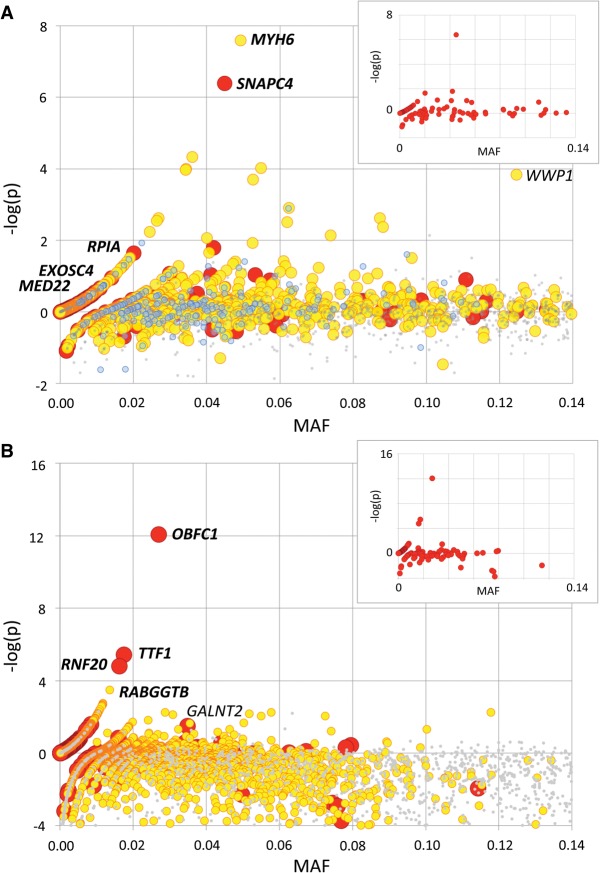
We herein report the result of a large-scale, next generation sequencing (NGS)-based screen for embryonic lethal (EL) mutations in Belgian beef and New Zealand dairy cattle. We estimated by simulation that cattle might carry, on average, ∼0.5 recessive EL mutations. We mined exome sequence data from >600 animals, and identified 1377 stop-gain, 3139 frame-shift, 1341 splice-site, 22,939 disruptive missense, 62,399 benign missense, and 92,163 synonymous variants. We show that cattle have a comparable load of loss-of-function (LoF) variants (defined as stop-gain, frame-shift, or splice-site variants) as humans despite having a more variable exome. We genotyped >40,000 animals for up to 296 LoF and 3483 disruptive missense, breed-specific variants. We identified candidate EL mutations based on the observation of a significant depletion in homozygotes. We estimated the proportion of EL mutations at 15% of tested LoF and 6% of tested disruptive missense variants. We confirmed the EL nature of nine candidate variants by genotyping 200 carrier × carrier trios, and demonstrating the absence of homozygous offspring. The nine identified EL mutations segregate at frequencies ranging from 1.2% to 6.6% in the studied populations and collectively account for the mortality of ∼0.6% of conceptuses. We show that EL mutations preferentially affect gene products fulfilling basic cellular functions. The resulting information will be useful to avoid at-risk matings, thereby improving fertility.
© 2016 Charlier et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources