

Filifactor alocis Promotes Neutrophil Degranulation and Chemotactic Activity
- PMID: 27647870
- PMCID: PMC5116720
- DOI: 10.1128/IAI.00496-16
Filifactor alocis Promotes Neutrophil Degranulation and Chemotactic Activity
Abstract
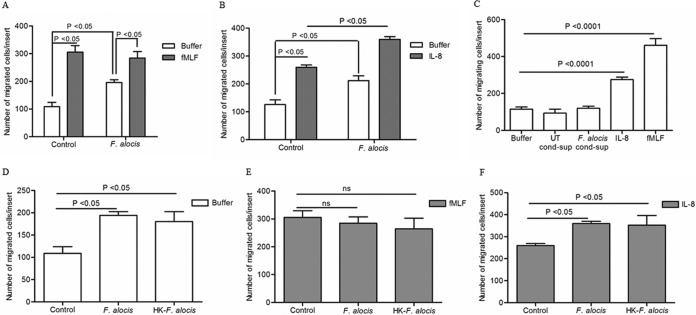
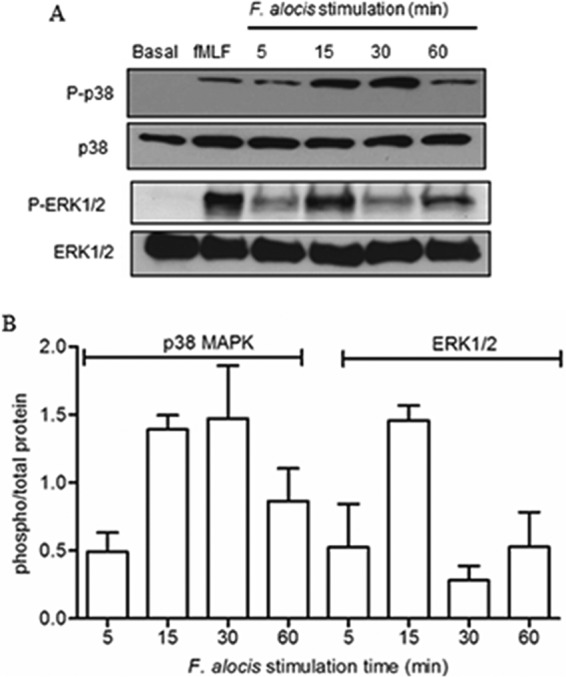
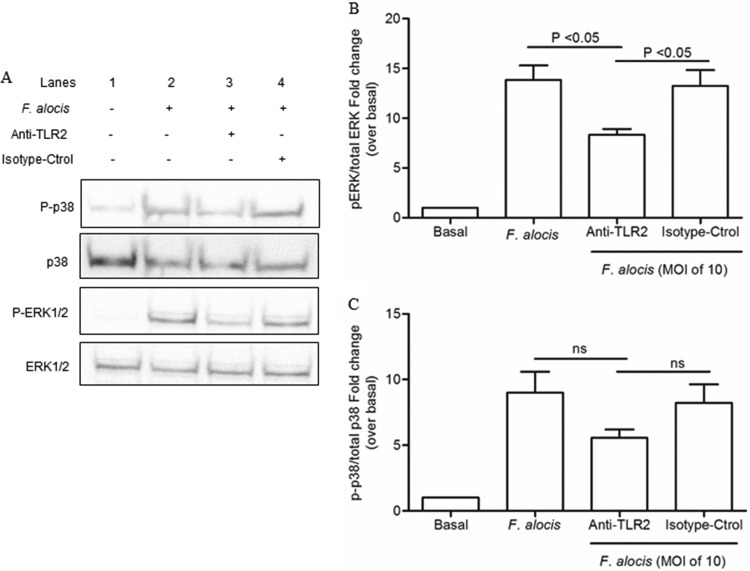
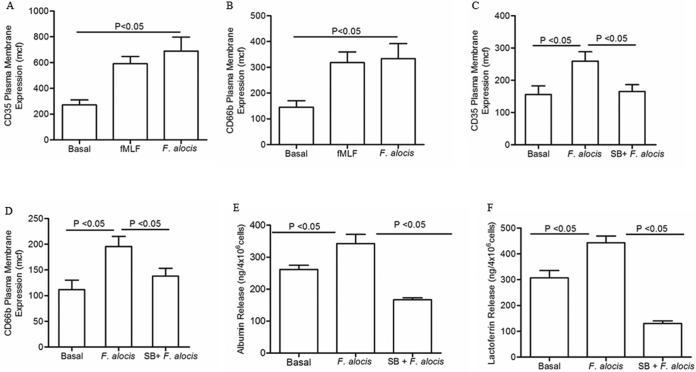
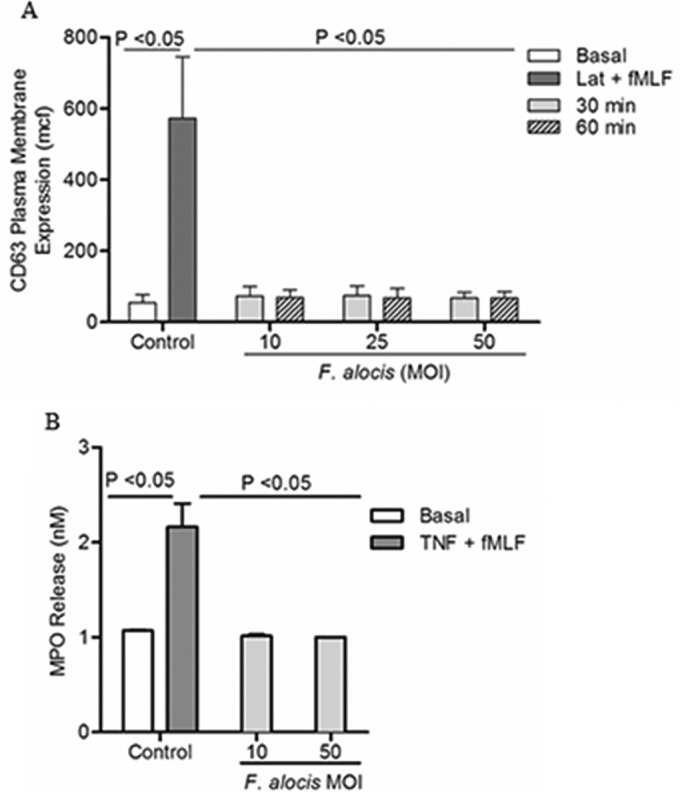
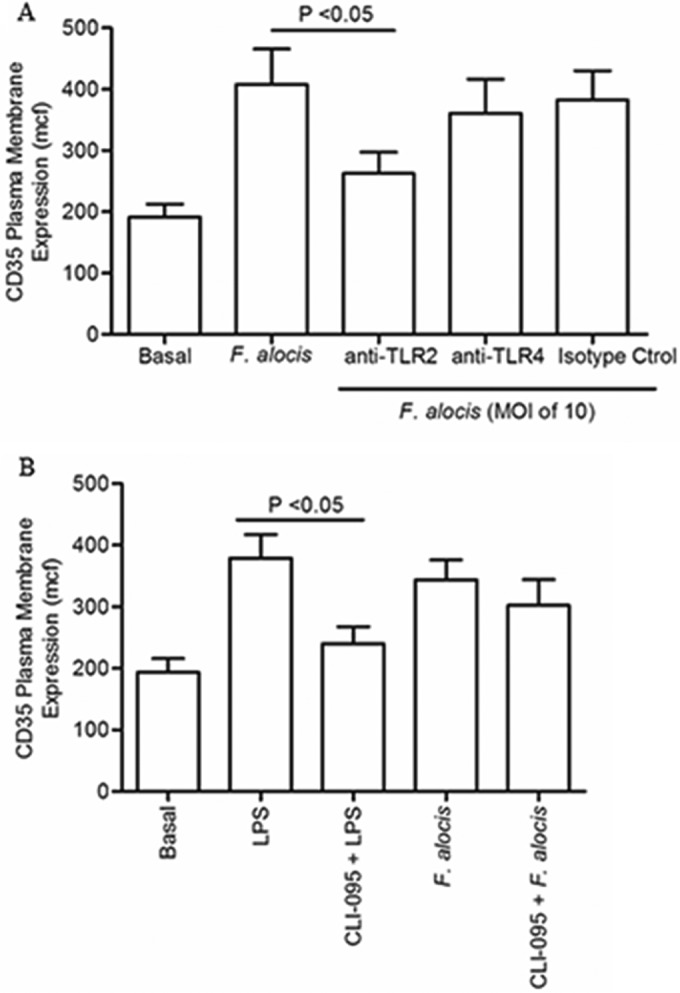
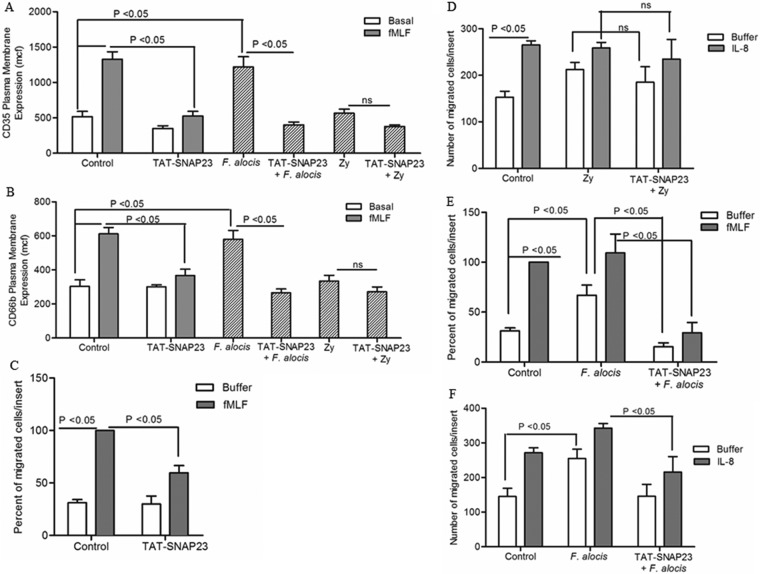
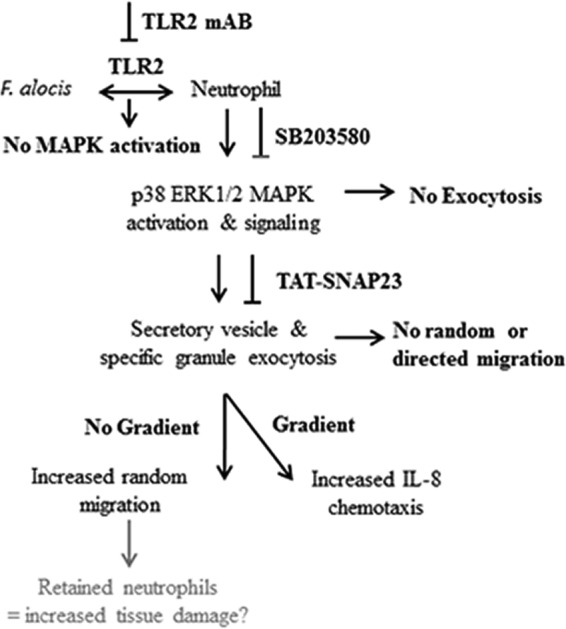
Filifactor alocis is a recently recognized periodontal pathogen; however, little is known regarding its interactions with the immune system. As the first-responder phagocytic cells, neutrophils are recruited in large numbers to the periodontal pocket, where they play a crucial role in the innate defense of the periodontium. Thus, in order to colonize, successful periodontal pathogens must devise means to interfere with neutrophil chemotaxis and activation. In this study, we assessed major neutrophil functions, including degranulation and cell migration, associated with the p38 mitogen-activated protein kinase (MAPK) signaling pathway upon challenge with F. alocis. Under conditions lacking a chemotactic gradient, F. alocis-challenged neutrophils had increased migration compared to uninfected cells, indicating that F. alocis increases chemokinesis in human neutrophils. In addition, neutrophil chemotaxis induced by interleukin-8 was significantly enhanced when cells were challenged with F. alocis, compared to noninfected cells. Similar to live bacteria, heat-killed F. alocis induced both random and directed migration of human neutrophils. The interaction of F. alocis with Toll-like receptor 2 induced granule exocytosis along with a transient ERK1/2 and sustained p38 MAPK activation. Moreover, F. alocis-induced secretory vesicle and specific granule exocytosis were p38 MAPK dependent. Blocking neutrophil degranulation with TAT-SNAP23 fusion protein significantly reduced the chemotactic and random migration induced by F. alocis Therefore, we propose that induction of random migration by F. alocis will prolong neutrophil traffic time in the gingival tissue, and subsequent degranulation will contribute to tissue damage.
Copyright © 2016, American Society for Microbiology. All Rights Reserved.
Figures
References
-
- Maddi A, Scannapieco FA. 2013. Oral biofilms, oral and periodontal infections, and systemic disease. Am J Dent 26:249–254. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
