

The DNA cytosine deaminase APOBEC3H haplotype I likely contributes to breast and lung cancer mutagenesis
- PMID: 27650891
- PMCID: PMC5036005
- DOI: 10.1038/ncomms12918
The DNA cytosine deaminase APOBEC3H haplotype I likely contributes to breast and lung cancer mutagenesis
Abstract
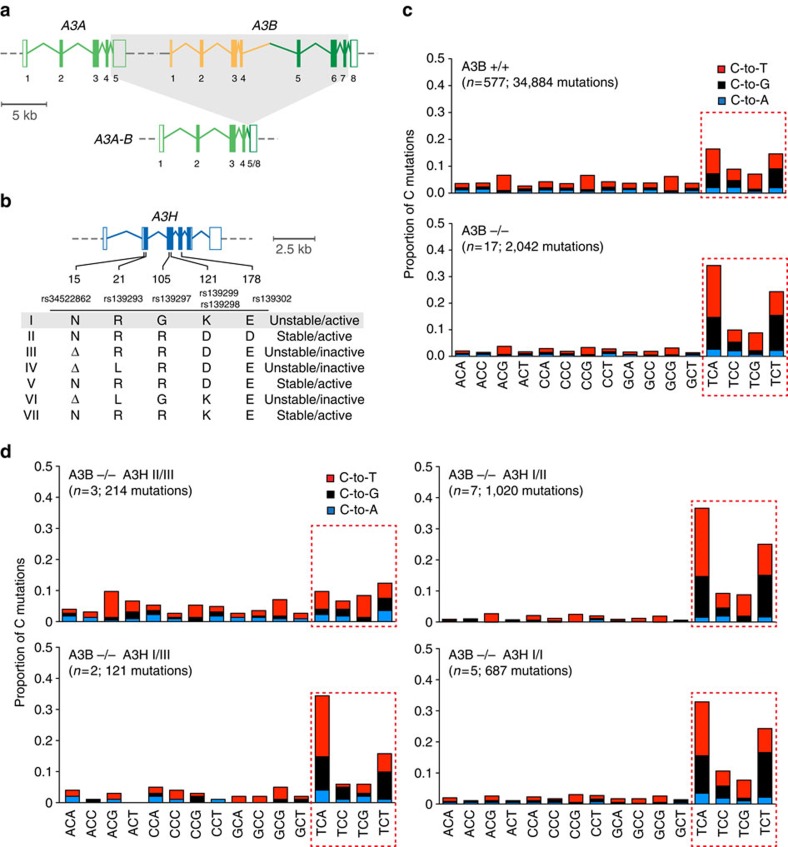
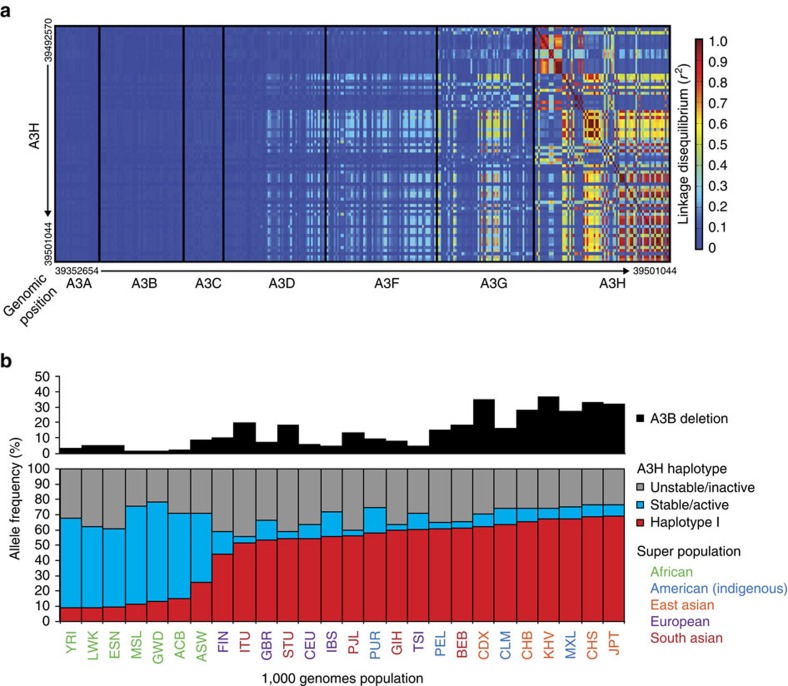
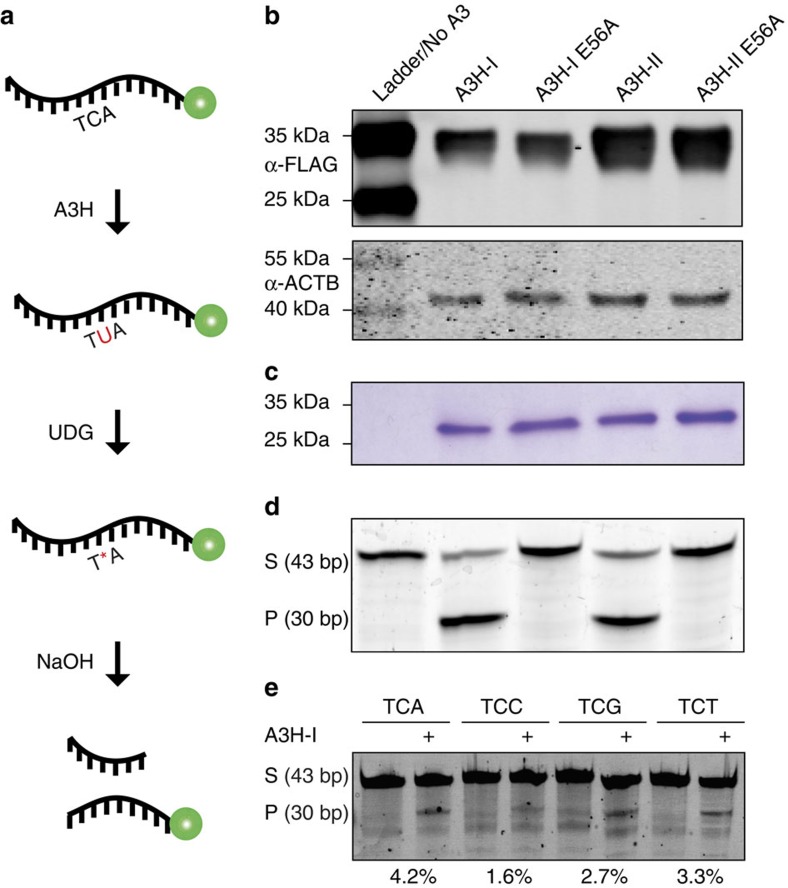
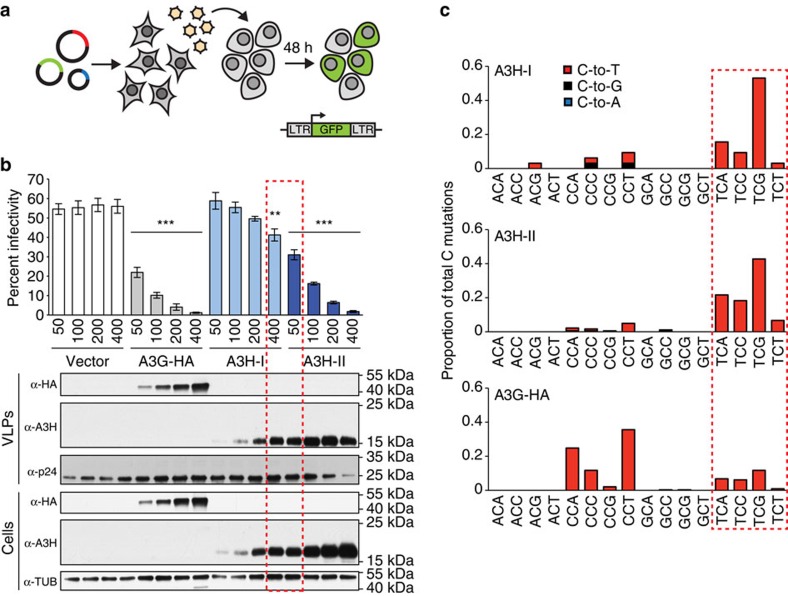
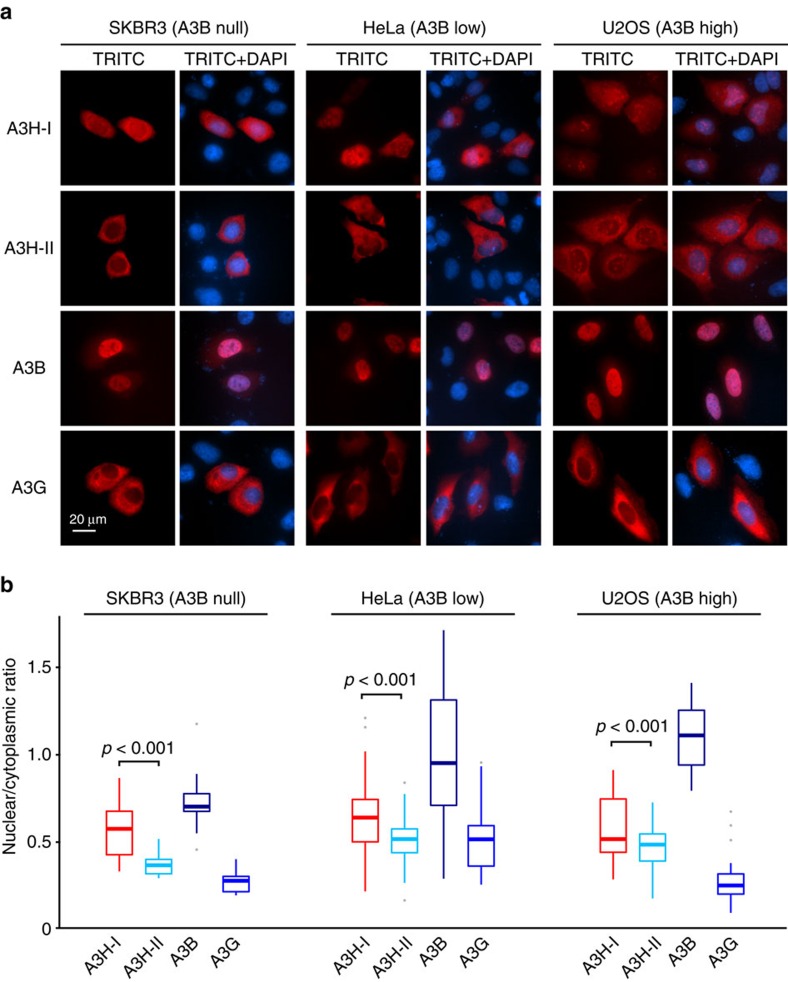
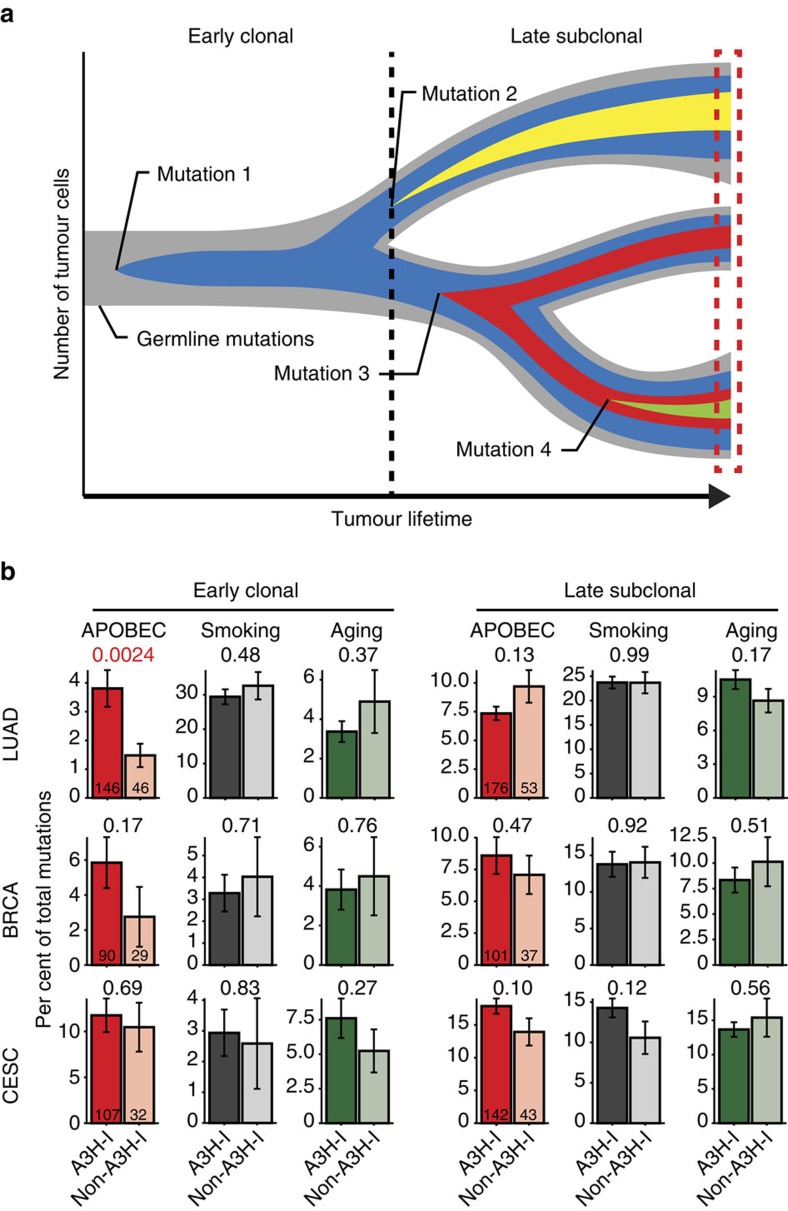
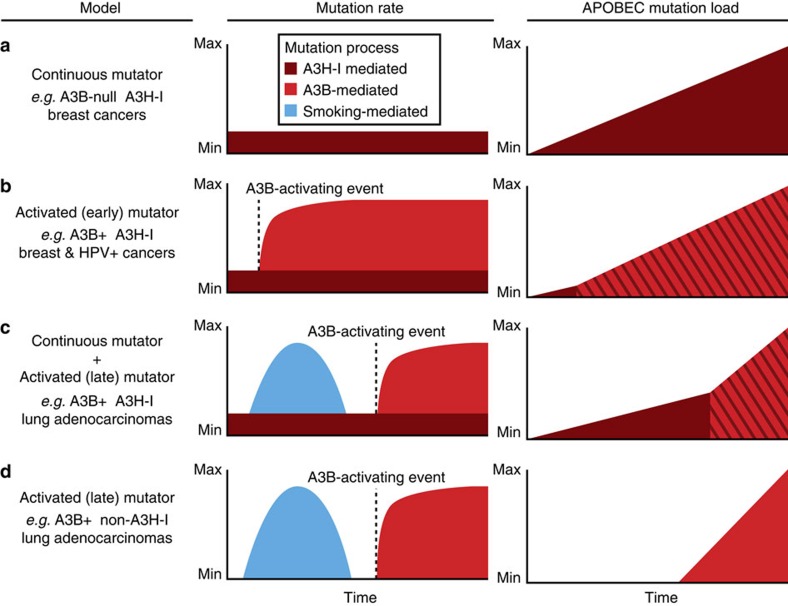
Cytosine mutations within TCA/T motifs are common in cancer. A likely cause is the DNA cytosine deaminase APOBEC3B (A3B). However, A3B-null breast tumours still have this mutational bias. Here we show that APOBEC3H haplotype I (A3H-I) provides a likely solution to this paradox. A3B-null tumours with this mutational bias have at least one copy of A3H-I despite little genetic linkage between these genes. Although deemed inactive previously, A3H-I has robust activity in biochemical and cellular assays, similar to A3H-II after compensation for lower protein expression levels. Gly105 in A3H-I (versus Arg105 in A3H-II) results in lower protein expression levels and increased nuclear localization, providing a mechanism for accessing genomic DNA. A3H-I also associates with clonal TCA/T-biased mutations in lung adenocarcinoma suggesting this enzyme makes broader contributions to cancer mutagenesis. These studies combine to suggest that A3B and A3H-I, together, explain the bulk of 'APOBEC signature' mutations in cancer.
Figures
References
-
- McGranahan N. & Swanton C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell 27, 15–26 (2015). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
