

A novel de novo TBX5 mutation in a patient with Holt-Oram syndrome leading to a dramatically reduced biological function
- PMID: 27652283
- PMCID: PMC5023941
- DOI: 10.1002/mgg3.234
A novel de novo TBX5 mutation in a patient with Holt-Oram syndrome leading to a dramatically reduced biological function
Abstract
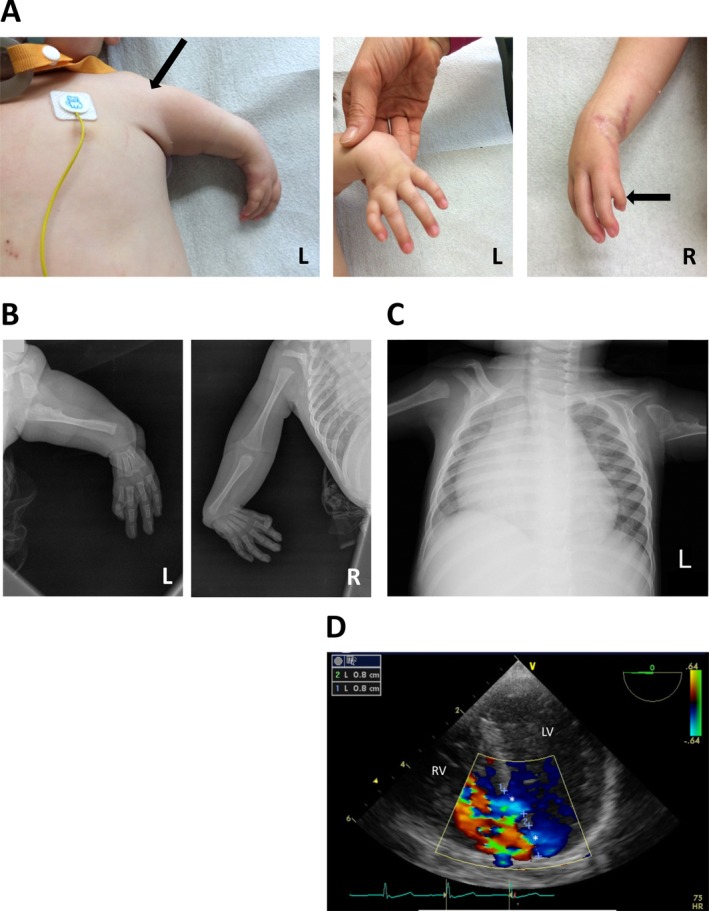
Background: The Holt-Oram syndrome (HOS) is an autosomal dominant disorder affecting 1/100.000 live births. It is defined by upper limb anomalies and congenital heart defects with variable severity. We describe a dramatic phenotype of a male, 15-month-old patient being investigated for strict diagnostic criteria of HOS.
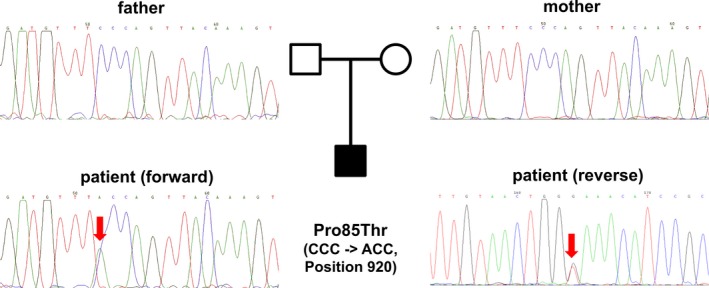
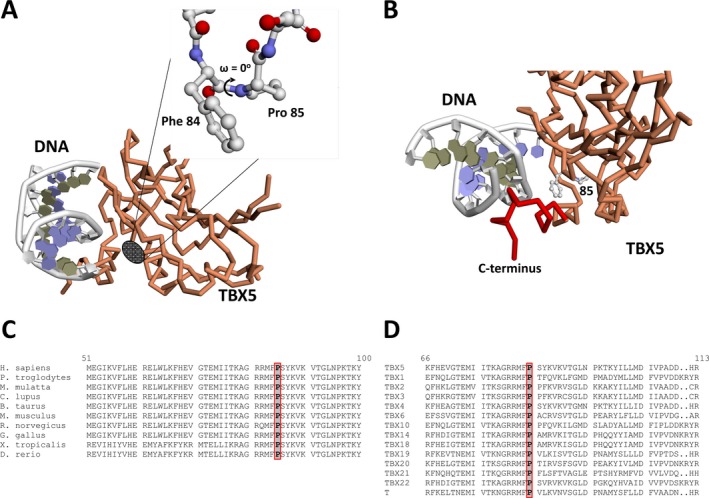
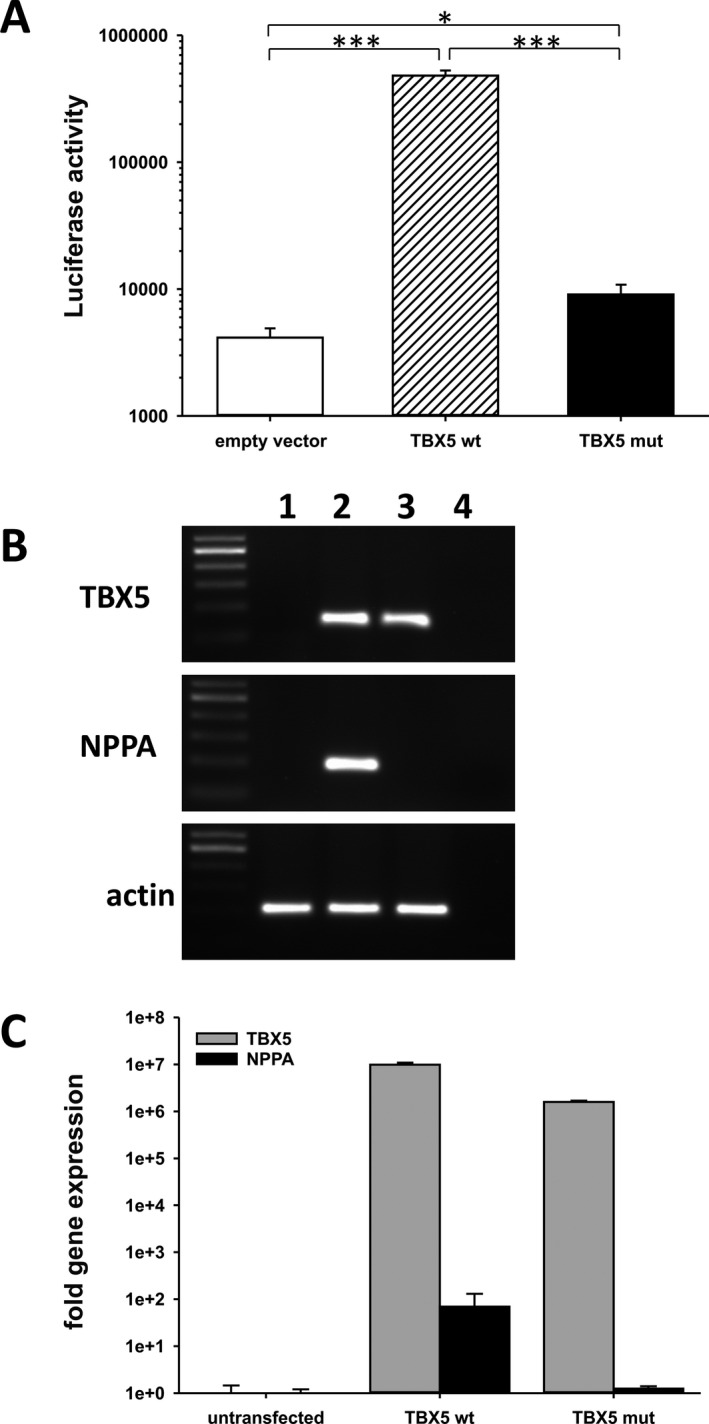
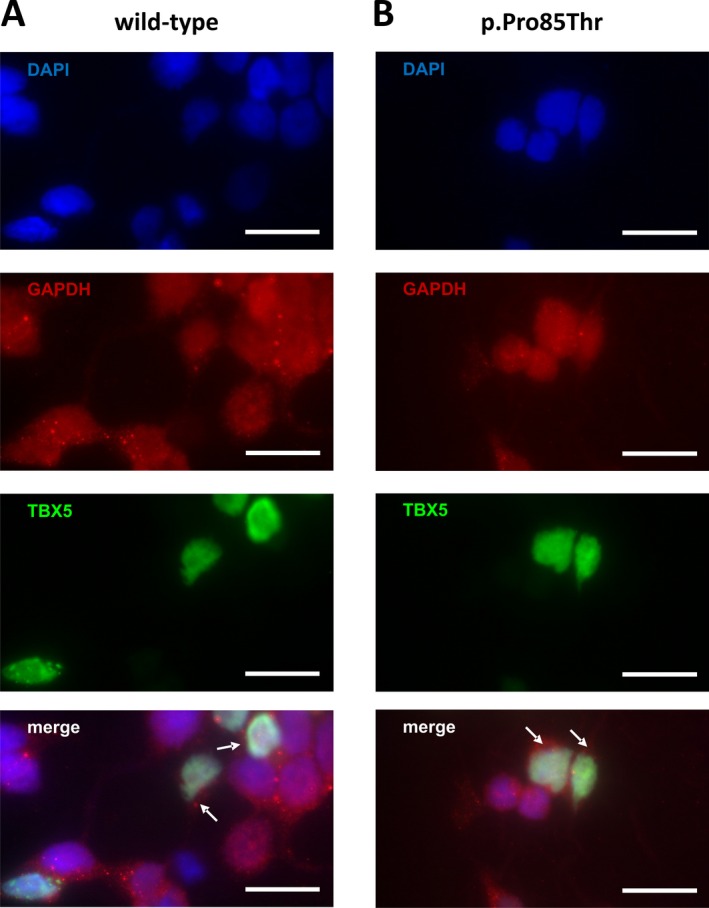
Methods and results: Genetic analysis revealed a so far unpublished TBX5 mutation, which occurs de novo in the patient with healthy parents. TBX5 belongs to the large family of T-box transcription factors playing major roles in morphogenesis and cell-type specification. The mutation located in the DNA-binding domain at position 920 (C→A) leads to an amino acid change at position 85 (proline → threonine). Three-dimensional analysis of the protein structure predicted a cis to trans change in the respective peptide bond, thereby probably provoking major conformational and functional alterations of the protein. The p.Pro85Thr mutation showed a dramatically reduced activation (97%) of the NPPA promoter in luciferase assays and failed to induce NPPA expression in HEK 293 cells compared to wild-type TBX5 protein. The mutation did not interfere with the nuclear localization of the protein.
Conclusion: These results suggest that the dramatic functional alteration of the p.Pro85Thr mutation leads to the distinctive phenotype of the patient.
Keywords: Congenital heart disease; Holt–Oram syndrome; TBX5; de novo mutation; heart‐hand syndrome; loss‐of function; transcription factor.
Figures
References
-
- Al‐Qattan, M. M. , and Abou Al‐Shaar H.. 2015. Molecular basis of the clinical features of Holt‐Oram syndrome resulting from missense and extended protein mutations of the TBX5 gene as well as TBX5 intragenic duplications. Gene 560:129–136. - PubMed
-
- Baban, A. , Postma A. V., Marini M., Trocchio G., Santilli A., Pelegrini M., et al. 2014. Identification of TBX5 mutations in a series of 94 patients with Tetralogy of Fallot. Am. J. Med. Genet. A 164A:3100–3107. - PubMed
-
- Bamshad, M. , Lin R. C., Law D. J., Watkins W. C., Krakowiak P. A., Moore M. E., et al. 1997. Mutations in human TBX3 alter limb, apocrine and genital development in ulnar‐mammary syndrome. Nat. Genet. 16:311–315. - PubMed
-
- Basson, C. T. , Cowley G. S., Solomon S. D., Weissman B., Poznanski A. K., Traill T. A., et al. 1994. The clinical and genetic spectrum of the Holt‐Oram syndrome (heart‐hand syndrome). N. Engl. J. Med. 330:885–891. - PubMed
-
- Basson, C. T. , Bachinsky D. R., Lin R. C., Levi T., Elkins J. A., Soults J., et al. 1997. Mutations in human TBX5 [corrected] cause limb and cardiac malformation in Holt‐Oram syndrome. Nat. Genet. 15:30–35. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
