

Targeted sequencing of 351 candidate genes for epileptic encephalopathy in a large cohort of patients
- PMID: 27652284
- PMCID: PMC5023942
- DOI: 10.1002/mgg3.235
Targeted sequencing of 351 candidate genes for epileptic encephalopathy in a large cohort of patients
Abstract
Background: Many genes are candidates for involvement in epileptic encephalopathy (EE) because one or a few possibly pathogenic variants have been found in patients, but insufficient genetic or functional evidence exists for a definite annotation.
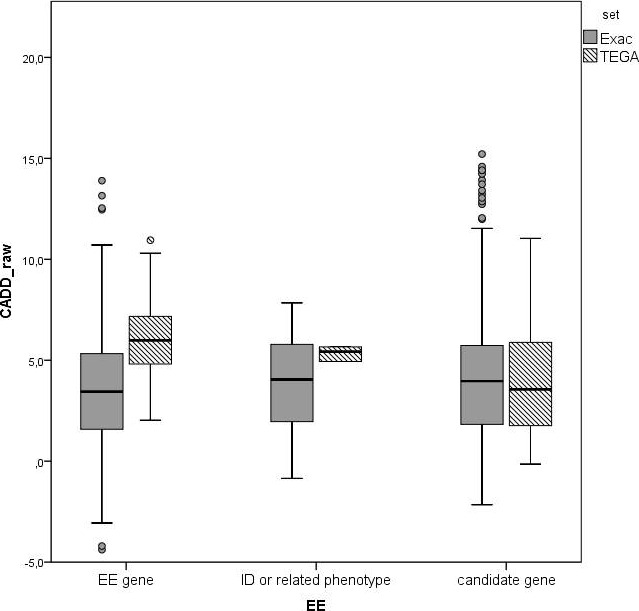
Methods: To increase the number of validated EE genes, we sequenced 26 known and 351 candidate genes for EE in 360 patients. Variants in 25 genes known to be involved in EE or related phenotypes were followed up in 41 patients. We prioritized the candidate genes, and followed up 31 variants in this prioritized subset of candidate genes.
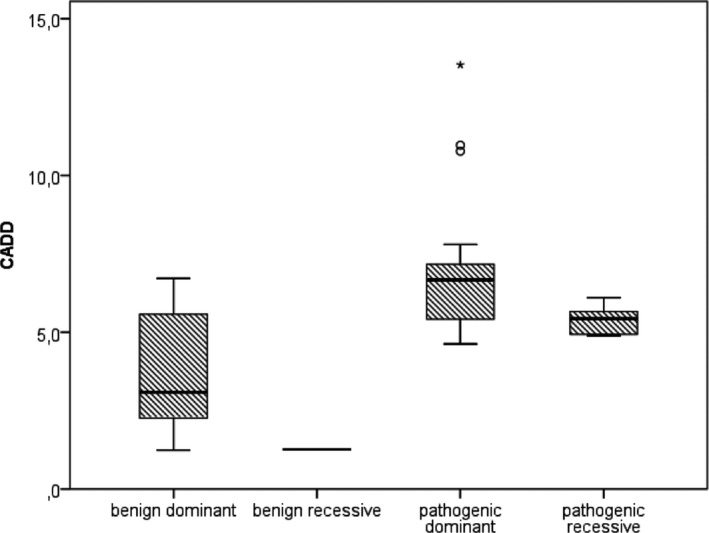
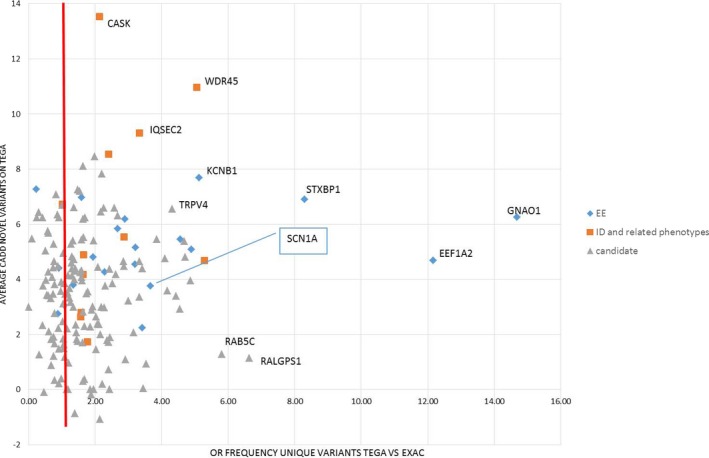
Results: Twenty-nine genotypes in known genes for EE (19) or related diseases (10), dominant as well as recessive or X-linked, were classified as likely pathogenic variants. Among those, likely pathogenic de novo variants were found in EE genes that act dominantly, including the recently identified genes EEF1A2, KCNB1 and the X-linked gene IQSEC2. A de novo frameshift variant in candidate gene HNRNPU was the only de novo variant found among the followed-up candidate genes, and the patient's phenotype was similar to a few recent publications.
Conclusion: Mutations in genes described in OMIM as, for example, intellectual disability gene can lead to phenotypes that get classified as EE in the clinic. We confirmed existing literature reports that de novo loss-of-function HNRNPUmutations lead to severe developmental delay and febrile seizures in the first year of life.
Keywords: De novo; HNRNPU; X‐linked; epileptic encephalopathy; loss‐of‐function; prioritization; recessive; targeted panel sequencing.
Figures
References
-
- Banne, E. , Atawneh O., Henneke M., Brockmann K., Gartner J., Elpeleg O., et al. 2013. West syndrome, microcephaly, grey matter heterotopia and hypoplasia of corpus callosum due to a novel ARFGEF2 mutation. J. Med. Genet. 50:772–775. - PubMed
-
- Caliebe, A. , Kroes H. Y., der van Smagt J. J., Martin‐Subero J. I., Tonnies H., van‘t Slot R. , et al. 2010. Four patients with speech delay, seizures and variable corpus callosum thickness sharing a 0.440 Mb deletion in region 1q44 containing the HNRPU gene. Eur. J. Med. Genet. 53:179–185. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
