

HIV-1 Escape from a Peptidic Anchor Inhibitor through Stabilization of the Envelope Glycoprotein Spike
- PMID: 27654295
- PMCID: PMC5110188
- DOI: 10.1128/JVI.01616-16
HIV-1 Escape from a Peptidic Anchor Inhibitor through Stabilization of the Envelope Glycoprotein Spike
Abstract
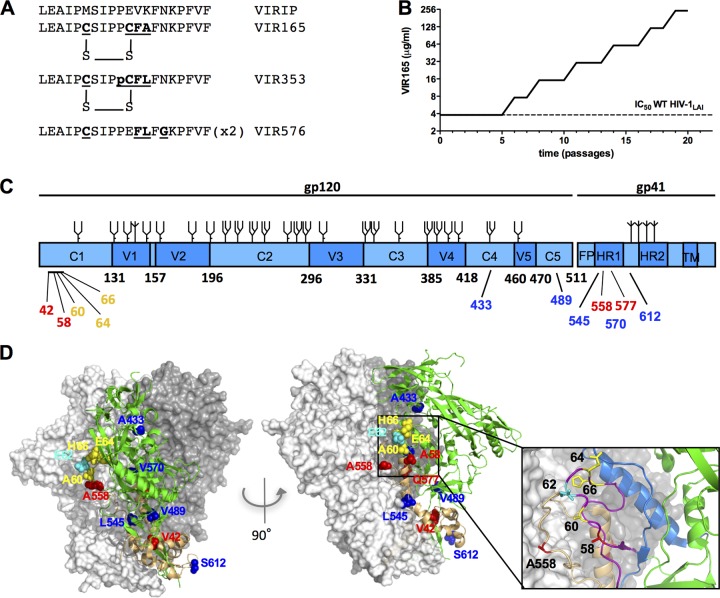
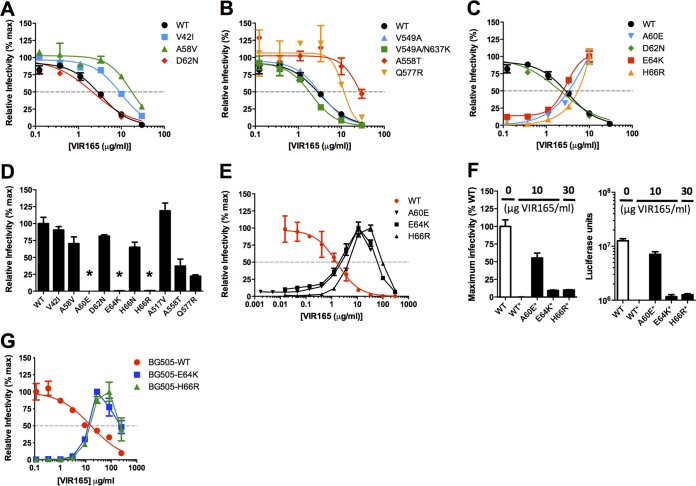
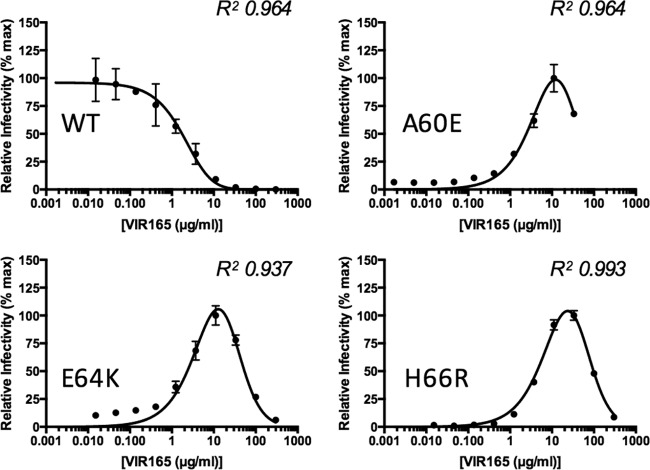
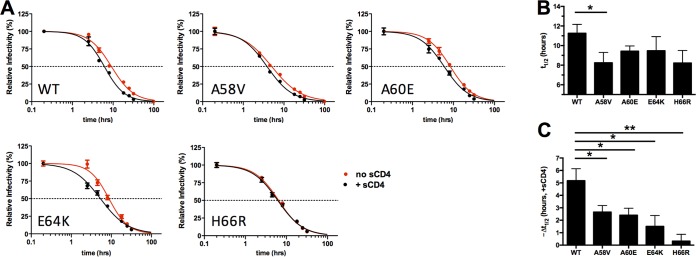
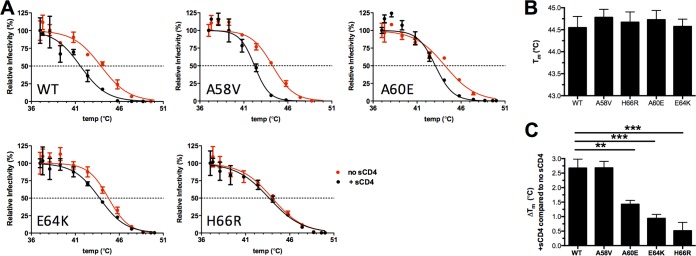
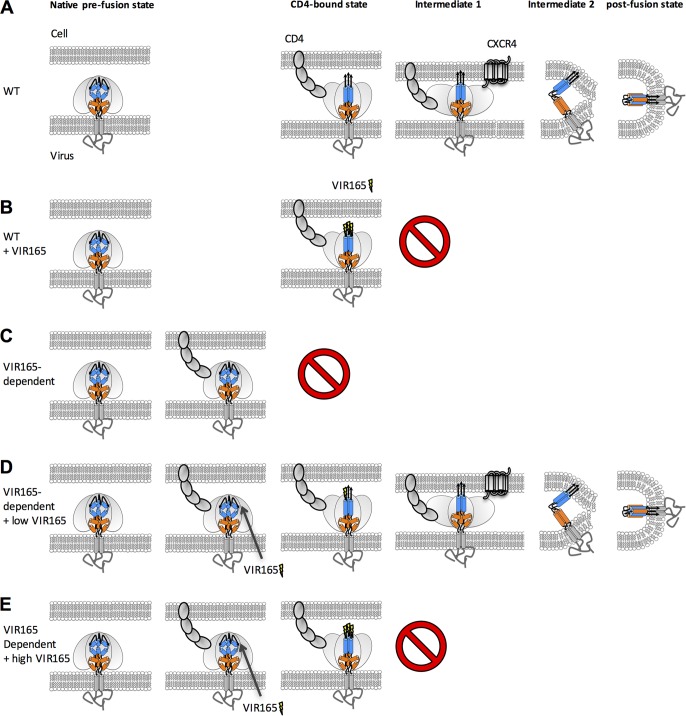
The trimeric HIV-1 envelope glycoprotein spike (Env) mediates viral entry into cells by using a spring-loaded mechanism that allows for the controlled insertion of the Env fusion peptide into the target membrane, followed by membrane fusion. Env is the focus of vaccine research aimed at inducing protective immunity by antibodies as well as efforts to develop drugs that inhibit the viral entry process. The molecular factors contributing to Env stability and decay need to be understood better in order to optimally design vaccines and therapeutics. We generated viruses with resistance to VIR165, a peptidic inhibitor that binds the fusion peptide of the gp41 subunit and prevents its insertion into the target membrane. Interestingly, a number of escape viruses acquired substitutions in the C1 domain of the gp120 subunit (A60E, E64K, and H66R) that rendered these viruses dependent on the inhibitor. These viruses could infect target cells only when VIR165 was present after CD4 binding. Furthermore, the VIR165-dependent viruses were resistant to soluble CD4-induced Env destabilization and decay. These data suggest that VIR165-dependent Env proteins are kinetically trapped in the unliganded state and require the drug to negotiate CD4-induced conformational changes. These studies provide mechanistic insight into the action of the gp41 fusion peptide and its inhibitors and provide new ways to stabilize Env trimer vaccines.
Importance: Because of the rapid development of HIV-1 drug resistance, new drug targets need to be explored continuously. The fusion peptide of the envelope glycoprotein can be targeted by anchor inhibitors. Here we describe virus escape from the anchor inhibitor VIR165. Interestingly, some escape viruses became dependent on the inhibitor for cell entry. We show that the identified escape mutations stabilize the ground state of the envelope glycoprotein and should thus be useful in the design of stabilized envelope-based HIV vaccines.
Copyright © 2016, American Society for Microbiology. All Rights Reserved.
Figures
Similar articles
-
Mutations That Increase the Stability of the Postfusion gp41 Conformation of the HIV-1 Envelope Glycoprotein Are Selected by both an X4 and R5 HIV-1 Virus To Escape Fusion Inhibitors Corresponding to Heptad Repeat 1 of gp41, but the gp120 Adaptive Mutations Differ between the Two Viruses.J Virol. 2019 May 15;93(11):e00142-19. doi: 10.1128/JVI.00142-19. Print 2019 Jun 1. J Virol. 2019. PMID: 30894471 Free PMC article.
-
HIV-1 gp41 Residues Modulate CD4-Induced Conformational Changes in the Envelope Glycoprotein and Evolution of a Relaxed Conformation of gp120.J Virol. 2018 Jul 31;92(16):e00583-18. doi: 10.1128/JVI.00583-18. Print 2018 Aug 15. J Virol. 2018. PMID: 29875245 Free PMC article.
-
Probing the Structure of the HIV-1 Envelope Trimer Using Aspartate Scanning Mutagenesis.J Virol. 2020 Oct 14;94(21):e01426-20. doi: 10.1128/JVI.01426-20. Print 2020 Oct 14. J Virol. 2020. PMID: 32817217 Free PMC article.
-
Quaternary Interaction of the HIV-1 Envelope Trimer with CD4 and Neutralizing Antibodies.Viruses. 2021 Jul 20;13(7):1405. doi: 10.3390/v13071405. Viruses. 2021. PMID: 34372611 Free PMC article. Review.
-
Development of Small-molecule HIV Entry Inhibitors Specifically Targeting gp120 or gp41.Curr Top Med Chem. 2016;16(10):1074-90. doi: 10.2174/1568026615666150901114527. Curr Top Med Chem. 2016. PMID: 26324044 Free PMC article. Review.
Cited by
-
HIV-1 anchor inhibitors and membrane fusion inhibitors target distinct but overlapping steps in virus entry.J Biol Chem. 2019 Apr 12;294(15):5736-5746. doi: 10.1074/jbc.RA119.007360. Epub 2019 Jan 29. J Biol Chem. 2019. PMID: 30696772 Free PMC article.
-
Identification of the natural product berberine as an antiviral drug.AMB Express. 2020 Sep 8;10(1):164. doi: 10.1186/s13568-020-01088-2. AMB Express. 2020. PMID: 32897426 Free PMC article.
-
Bifunctional Chimera That Coordinately Targets Human Immunodeficiency Virus 1 Envelope gp120 and the Host-Cell CCR5 Coreceptor at the Virus-Cell Interface.J Med Chem. 2018 Jun 14;61(11):5020-5033. doi: 10.1021/acs.jmedchem.8b00477. Epub 2018 Jun 1. J Med Chem. 2018. PMID: 29767965 Free PMC article.
-
A Virion-Based Assay for Glycoprotein Thermostability Reveals Key Determinants of Filovirus Entry and Its Inhibition.J Virol. 2020 Aug 31;94(18):e00336-20. doi: 10.1128/JVI.00336-20. Print 2020 Aug 31. J Virol. 2020. PMID: 32611759 Free PMC article.
-
Functional Stability of HIV-1 Envelope Trimer Affects Accessibility to Broadly Neutralizing Antibodies at Its Apex.J Virol. 2017 Nov 30;91(24):e01216-17. doi: 10.1128/JVI.01216-17. Print 2017 Dec 15. J Virol. 2017. PMID: 28978711 Free PMC article.
References
-
- Kong R, Xu K, Zhou T, Acharya P, Lemmin T, Liu K, Ozorowski G, Soto C, Taft JD, Bailer RT, Cale EM, Chen L, Choi CW, Chuang GY, Doria-Rose NA, Druz A, Georgiev IS, Gorman J, Huang J, Joyce MG, Louder MK, Ma X, McKee K, O'Dell S, Pancera M, Yang Y, Blanchard SC, Mothes W, Burton DR, Koff WC, Connors M, Ward AB, Kwong PD, Mascola JR. 2016. Fusion peptide of HIV-1 as a site of vulnerability to neutralizing antibody. Science 352:828–833. doi:10.1126/science.aae0474. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
