

A genomic history of Aboriginal Australia
- PMID: 27654914
- PMCID: PMC7617037
- DOI: 10.1038/nature18299
A genomic history of Aboriginal Australia
Abstract
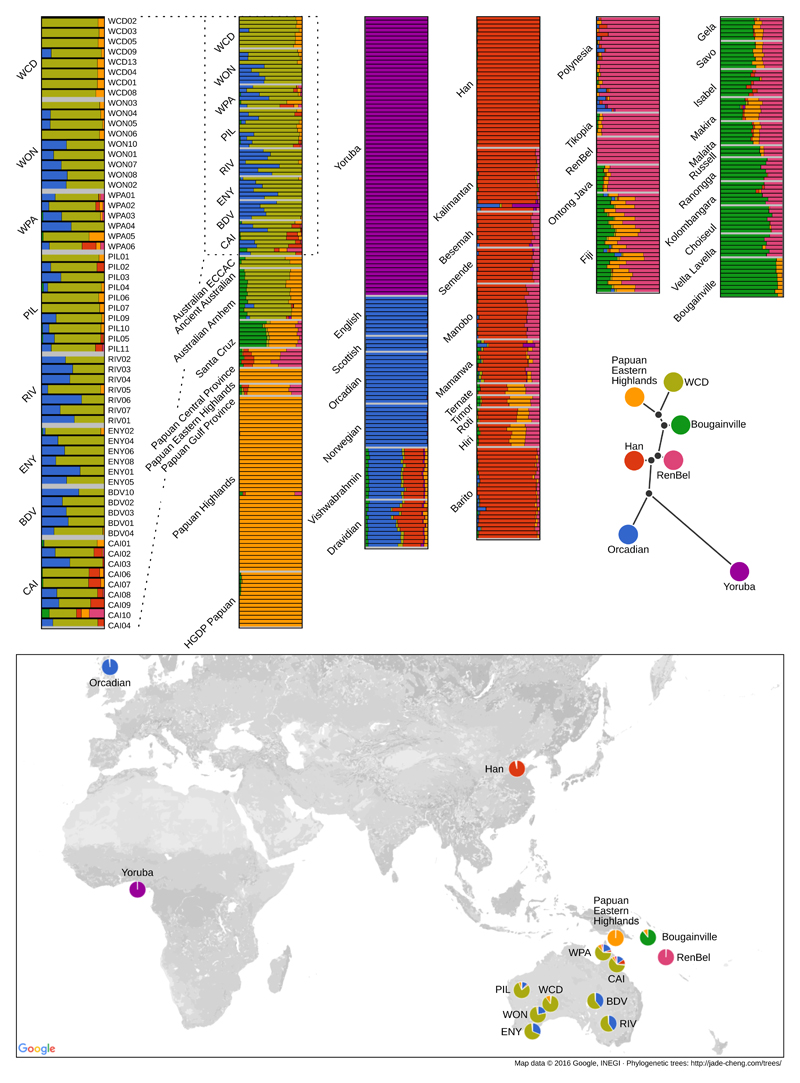
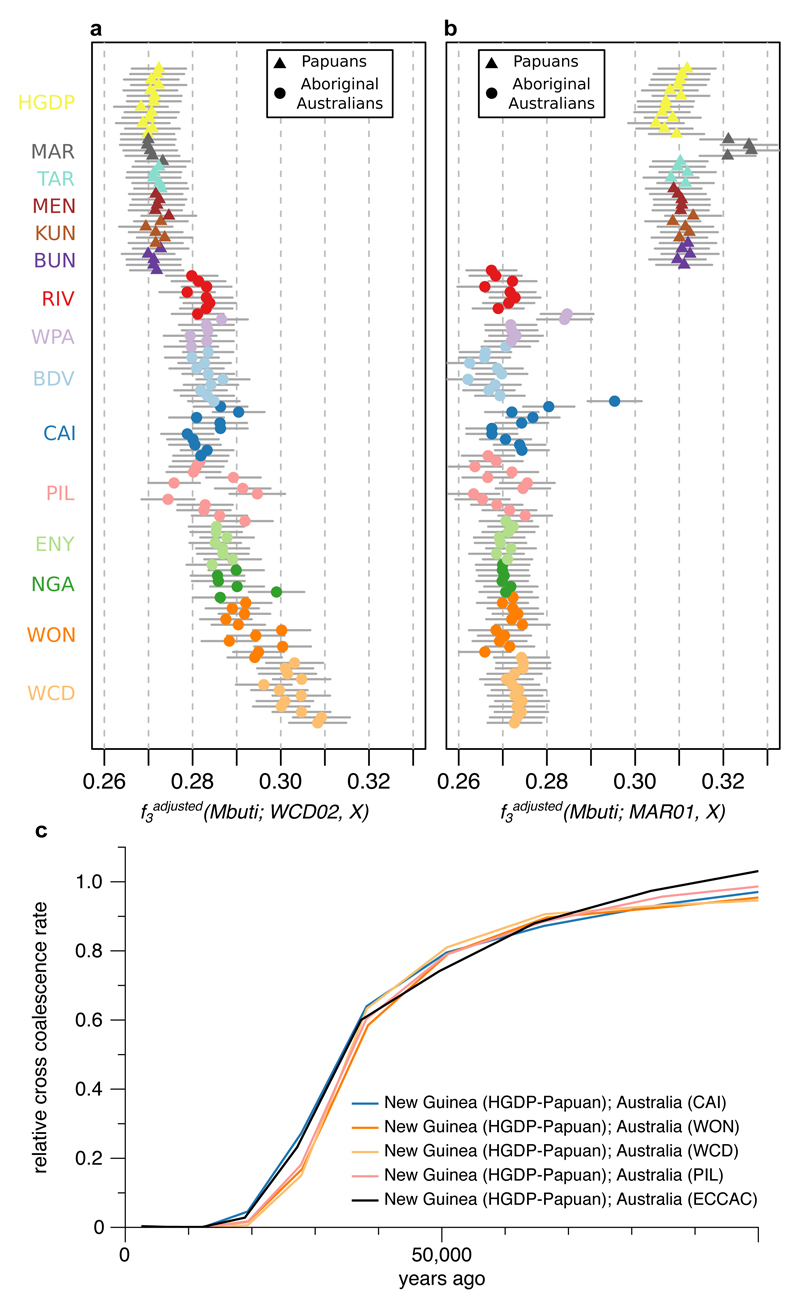
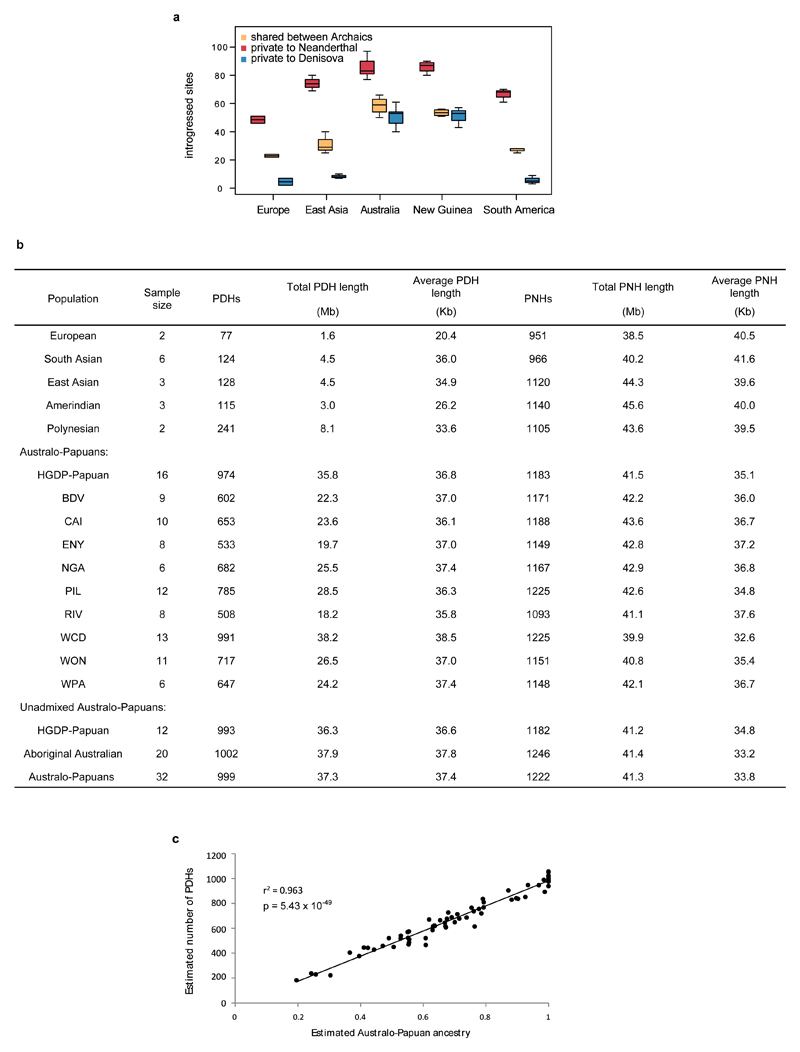
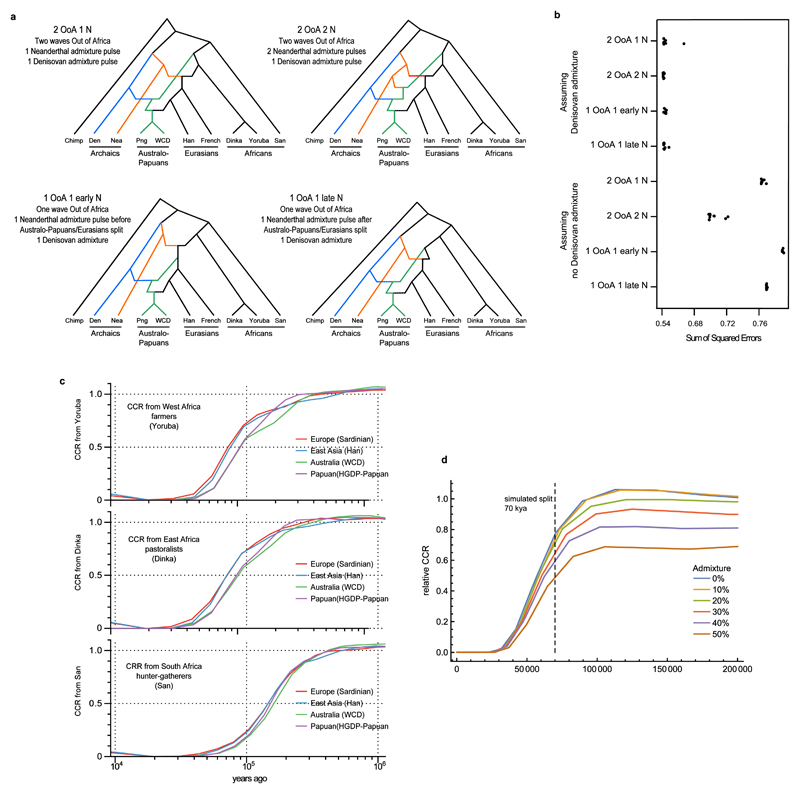
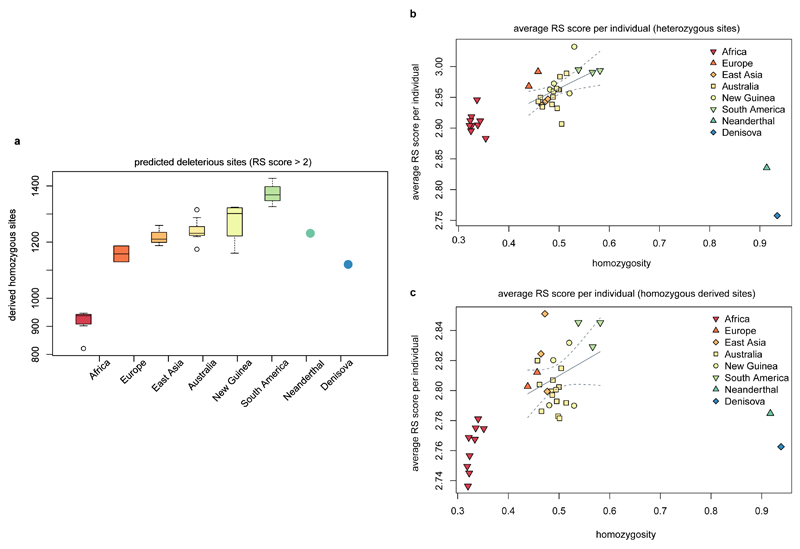
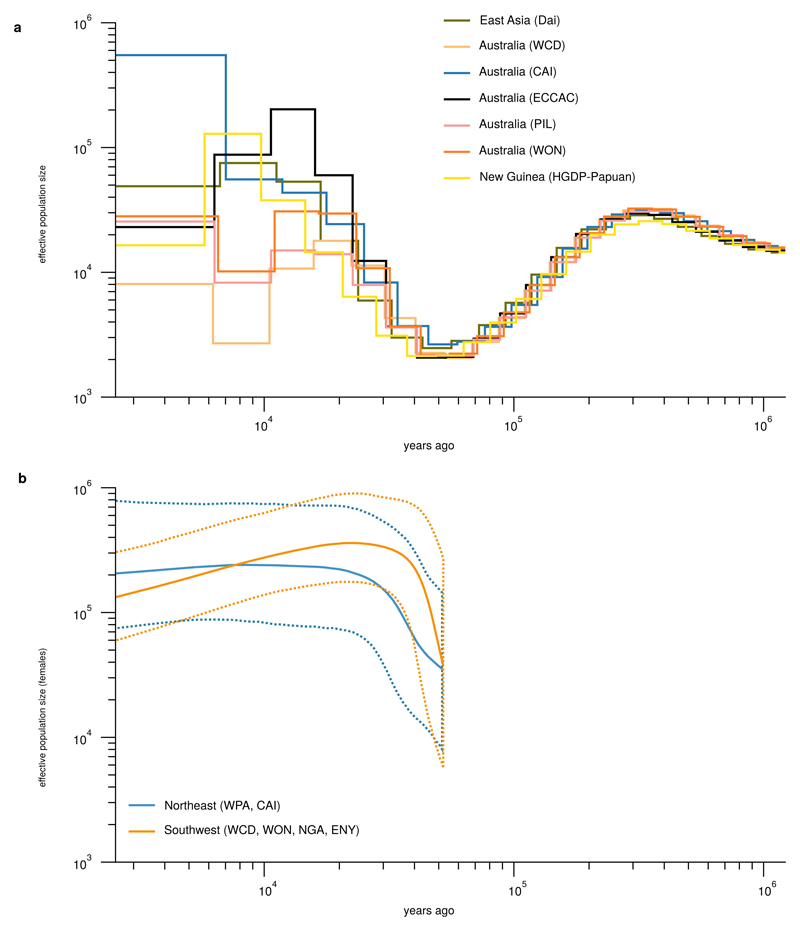
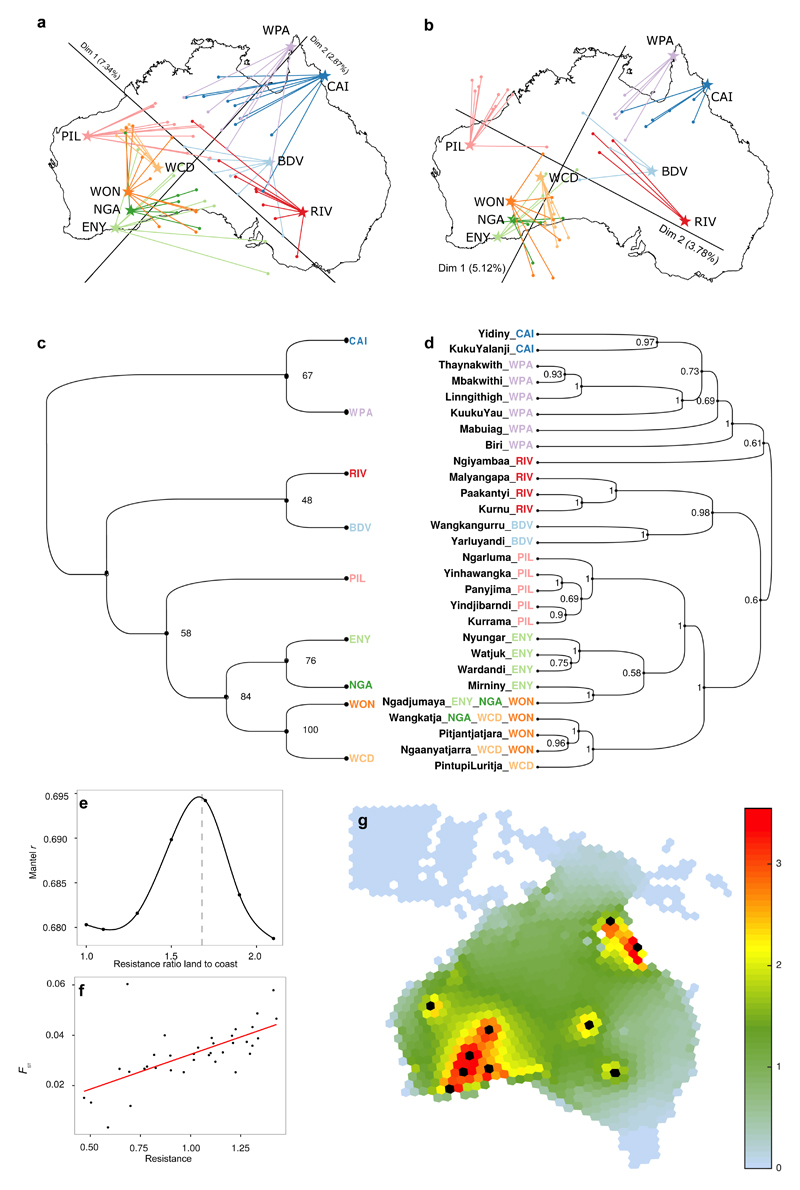
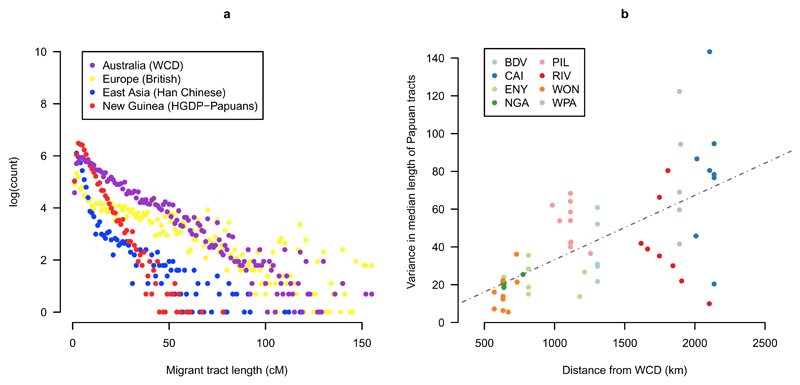
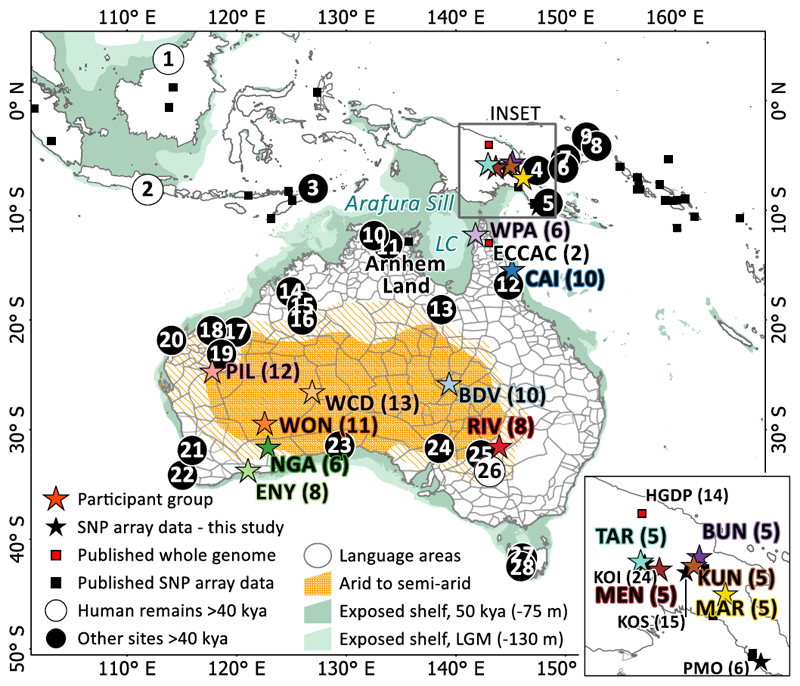
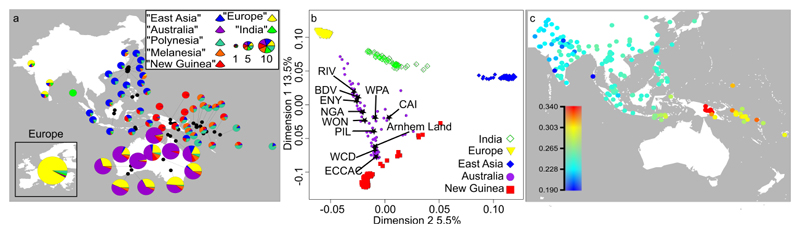
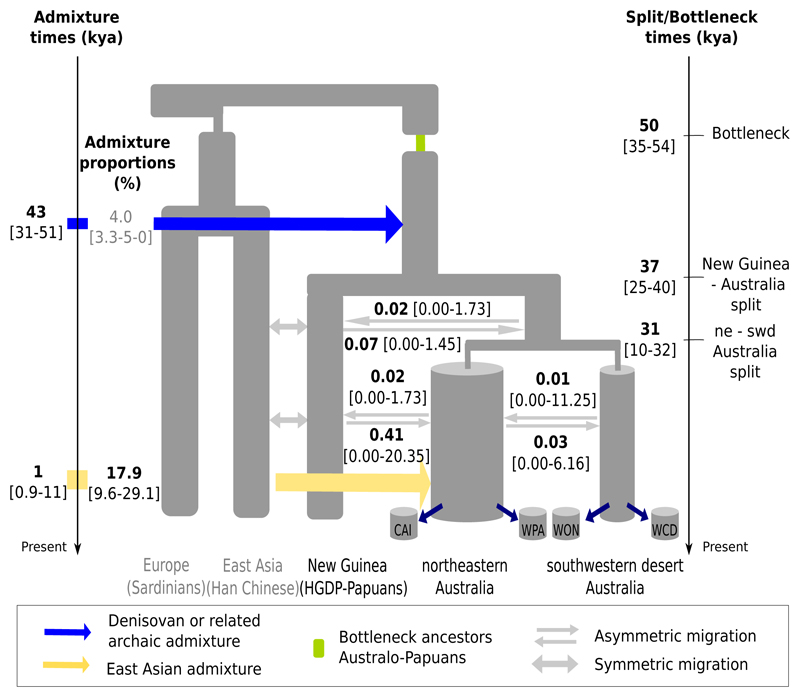
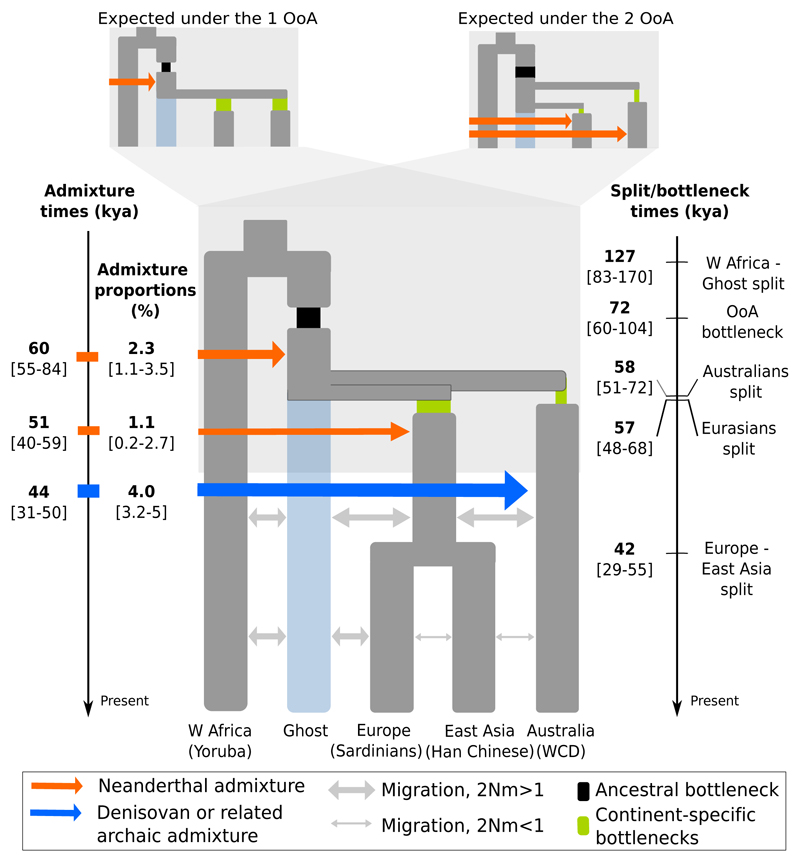
The population history of Aboriginal Australians remains largely uncharacterized. Here we generate high-coverage genomes for 83 Aboriginal Australians (speakers of Pama-Nyungan languages) and 25 Papuans from the New Guinea Highlands. We find that Papuan and Aboriginal Australian ancestors diversified 25-40 thousand years ago (kya), suggesting pre-Holocene population structure in the ancient continent of Sahul (Australia, New Guinea and Tasmania). However, all of the studied Aboriginal Australians descend from a single founding population that differentiated ~10-32 kya. We infer a population expansion in northeast Australia during the Holocene epoch (past 10,000 years) associated with limited gene flow from this region to the rest of Australia, consistent with the spread of the Pama-Nyungan languages. We estimate that Aboriginal Australians and Papuans diverged from Eurasians 51-72 kya, following a single out-of-Africa dispersal, and subsequently admixed with archaic populations. Finally, we report evidence of selection in Aboriginal Australians potentially associated with living in the desert.
Figures
Comment in
-
Geneticists attempt to heal rifts with Aboriginal communities.Nature. 2016 Sep 22;537(7621):457-8. doi: 10.1038/537457a. Nature. 2016. PMID: 27652541 No abstract available.
-
Population genetics: A map of human wanderlust.Nature. 2016 Oct 13;538(7624):179-180. doi: 10.1038/nature19472. Epub 2016 Sep 21. Nature. 2016. PMID: 27654916 No abstract available.
-
Consent: Data-sharing for indigenous peoples.Nature. 2017 Jun 21;546(7659):474. doi: 10.1038/546474a. Nature. 2017. PMID: 28640268 No abstract available.
-
Data sharing: do scientists know best?Nature. 2017 Aug 16;548(7667):281. doi: 10.1038/548281b. Nature. 2017. PMID: 28816250 No abstract available.
References
-
- Davidson I. The colonization of Australia and its adjacent islands and the evolution of modern cognition. Curr Anthropol. 2010;51:S177–S189.
-
- Clarkson C, et al. The archaeology, chronology and stratigraphy of Madjedbebe (Malakunanja II): A site in northern Australia with early occupation. J Hum Evol. 2015;83:46–64. - PubMed
-
- O’Connell JF, Allen J. The process, biotic impact, and global implications of the human colonization of Sahul about 47,000 years ago. J Archaeol Sci. 2015;56:73–84.
-
- Barker G, et al. The ‘human revolution’in tropical Southeast Asia: the antiquity of anatomically modern humans, and of behavioural modernity, at Niah Cave (Sarawak, Borneo) J Hum Evol. 2007;52:243–261. - PubMed
-
- Lahr MM, Foley R. Multiple dispersals and modern human origins. Evol Anthropol Issues News Rev. 1994;3:48–60.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
