

Incorporating Functional Annotations for Fine-Mapping Causal Variants in a Bayesian Framework Using Summary Statistics
- PMID: 27655946
- PMCID: PMC5105870
- DOI: 10.1534/genetics.116.188953
Incorporating Functional Annotations for Fine-Mapping Causal Variants in a Bayesian Framework Using Summary Statistics
Abstract
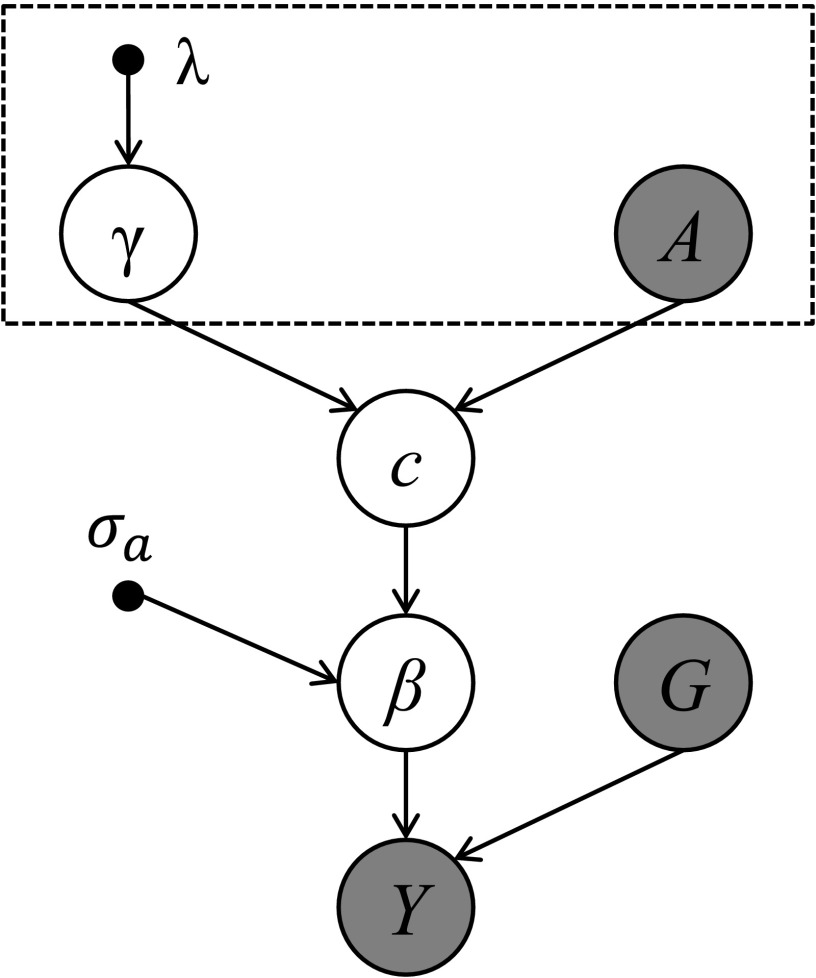
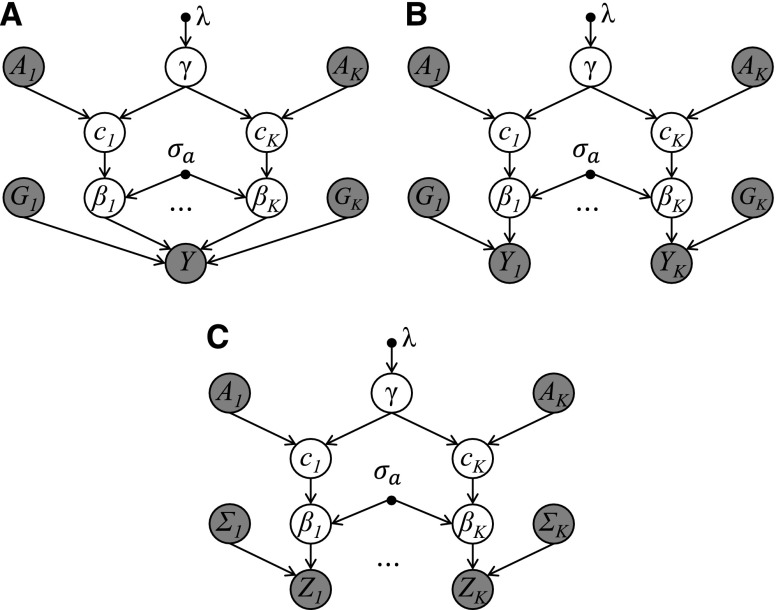
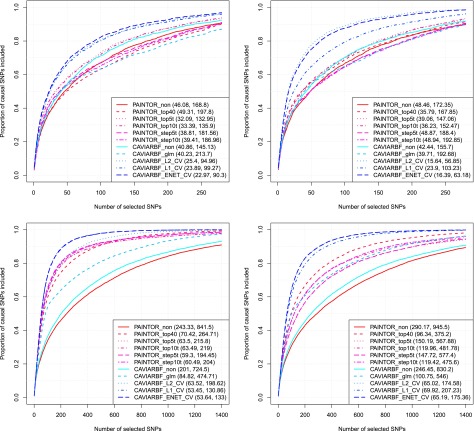
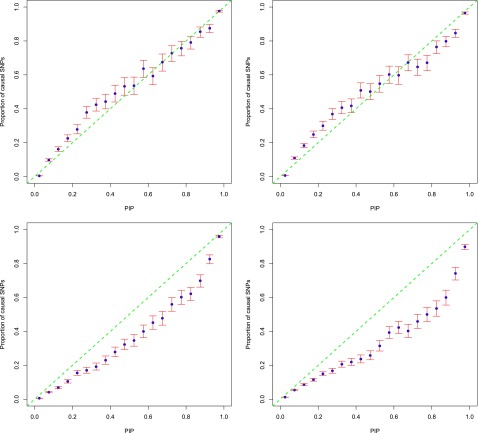
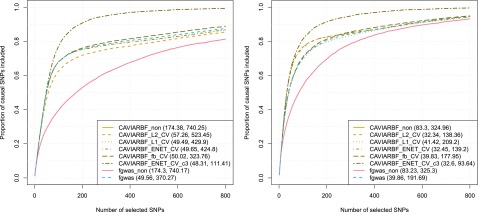
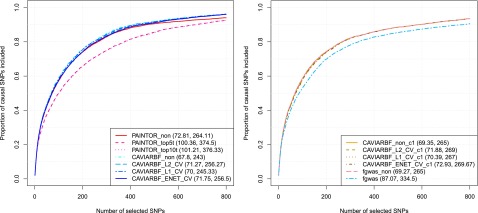
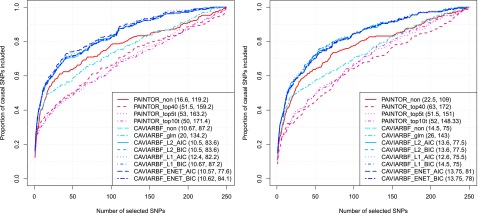
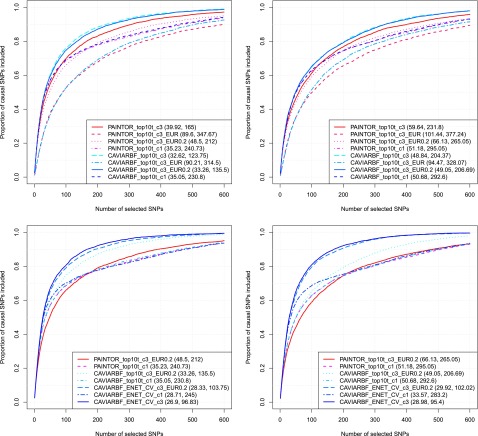
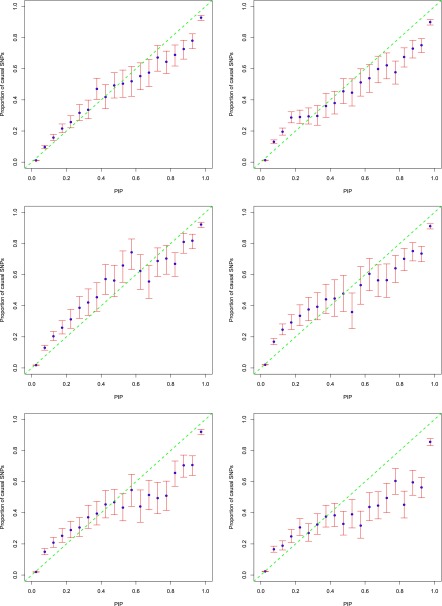
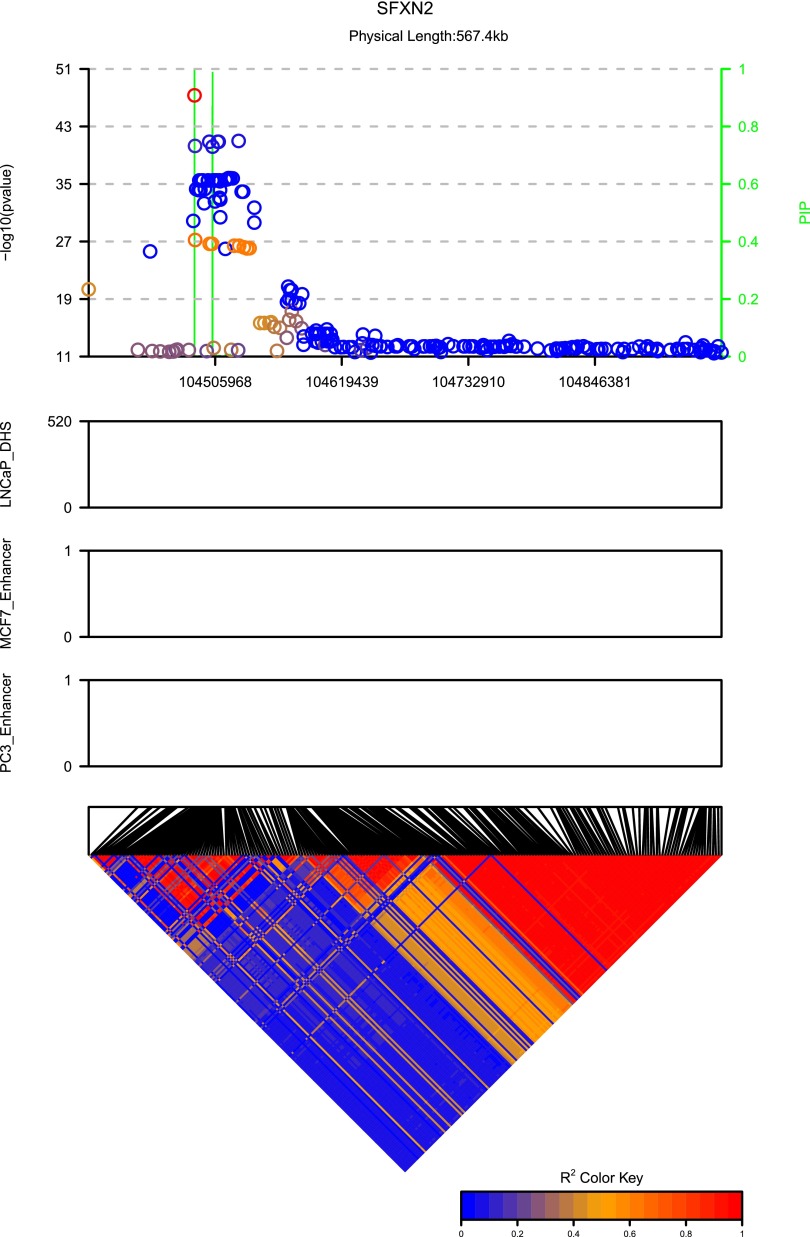
Functional annotations have been shown to improve both the discovery power and fine-mapping accuracy in genome-wide association studies. However, the optimal strategy to incorporate the large number of existing annotations is still not clear. In this study, we propose a Bayesian framework to incorporate functional annotations in a systematic manner. We compute the maximum a posteriori solution and use cross validation to find the optimal penalty parameters. By extending our previous fine-mapping method CAVIARBF into this framework, we require only summary statistics as input. We also derived an exact calculation of Bayes factors using summary statistics for quantitative traits, which is necessary when a large proportion of trait variance is explained by the variants of interest, such as in fine mapping expression quantitative trait loci (eQTL). We compared the proposed method with PAINTOR using different strategies to combine annotations. Simulation results show that the proposed method achieves the best accuracy in identifying causal variants among the different strategies and methods compared. We also find that for annotations with moderate effects from a large annotation pool, screening annotations individually and then combining the top annotations can produce overly optimistic results. We applied these methods on two real data sets: a meta-analysis result of lipid traits and a cis-eQTL study of normal prostate tissues. For the eQTL data, incorporating annotations significantly increased the number of potential causal variants with high probabilities.
Keywords: Bayesian fine mapping; annotations; causal variants; summary statistics.
Copyright © 2016 by the Genetics Society of America.
Figures
References
-
- Bert S. A., Robinson M. D., Strbenac D., Statham A. L., Song J. Z., et al. , 2013. Regional activation of the cancer genome by long-range epigenetic remodeling. Cancer Cell 23: 9–22. - PubMed
-
- Bishop C. M., 2006. Pattern Recognition and Machine Learning. Springer, New York.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
