

CFTR Modulators: Shedding Light on Precision Medicine for Cystic Fibrosis
- PMID: 27656143
- PMCID: PMC5011145
- DOI: 10.3389/fphar.2016.00275
CFTR Modulators: Shedding Light on Precision Medicine for Cystic Fibrosis
Abstract
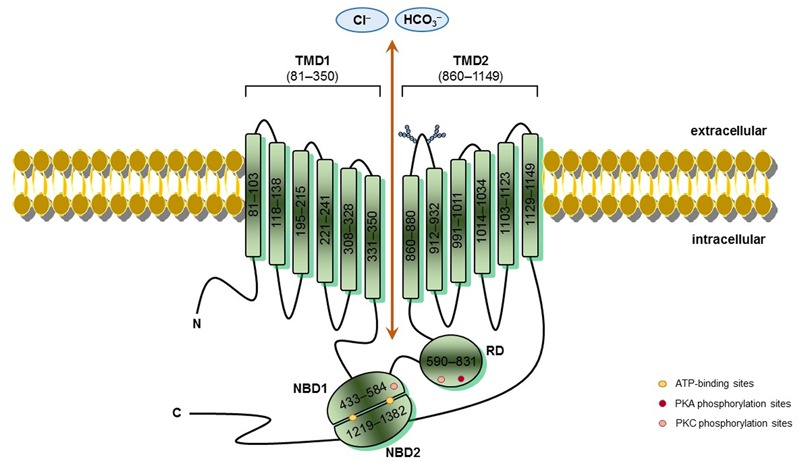
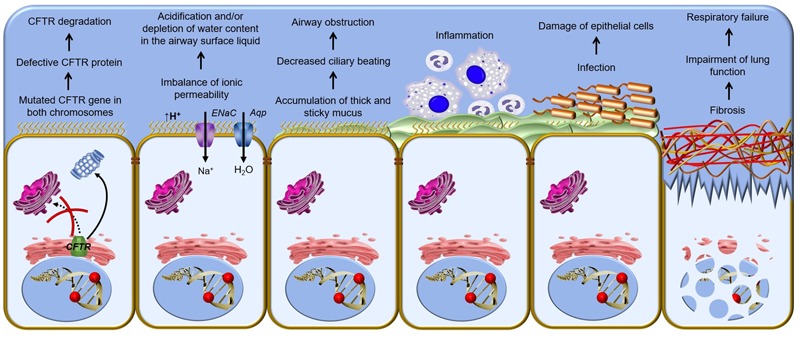
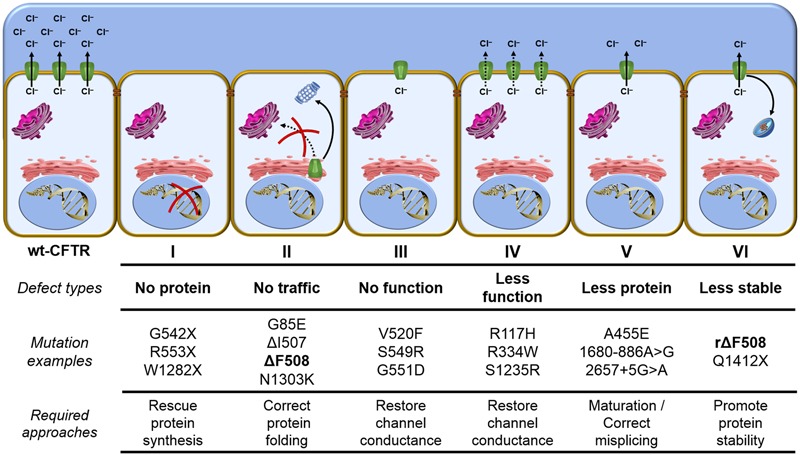
Cystic fibrosis (CF) is the most common life-threatening monogenic disease afflicting Caucasian people. It affects the respiratory, gastrointestinal, glandular and reproductive systems. The major cause of morbidity and mortality in CF is the respiratory disorder caused by a vicious cycle of obstruction of the airways, inflammation and infection that leads to epithelial damage, tissue remodeling and end-stage lung disease. Over the past decades, life expectancy of CF patients has increased due to early diagnosis and improved treatments; however, these patients still present limited quality of life. Many attempts have been made to rescue CF transmembrane conductance regulator (CFTR) expression, function and stability, thereby overcoming the molecular basis of CF. Gene and protein variances caused by CFTR mutants lead to different CF phenotypes, which then require different treatments to quell the patients' debilitating symptoms. In order to seek better approaches to treat CF patients and maximize therapeutic effects, CFTR mutants have been stratified into six groups (although several of these mutations present pleiotropic defects). The research with CFTR modulators (read-through agents, correctors, potentiators, stabilizers and amplifiers) has achieved remarkable progress, and these drugs are translating into pharmaceuticals and personalized treatments for CF patients. This review summarizes the main molecular and clinical features of CF, emphasizes the latest clinical trials using CFTR modulators, sheds light on the molecular mechanisms underlying these new and emerging treatments, and discusses the major breakthroughs and challenges to treating all CF patients.
Keywords: ABC transporters; CFTR; cystic fibrosis; intracellular trafficking; personalized medicine; protein misfolding; proteostasis network.
Figures
References
-
- Ahner A., Gong X., Schmidt B. Z., Peters K. W., Rabeh W. M., Thibodeau P. H., et al. (2013). Small heat shock proteins target mutant cystic fibrosis transmembrane conductance regulator for degradation via a small ubiquitin-like modifier-dependent pathway. Mol. Biol. Cell 24 74–84. 10.1091/mbc.E12-09-0678 - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous