

The validation of pharmacogenetics for the identification of Fabry patients to be treated with migalastat
- PMID: 27657681
- PMCID: PMC5392595
- DOI: 10.1038/gim.2016.122
The validation of pharmacogenetics for the identification of Fabry patients to be treated with migalastat
Abstract
Purpose: Fabry disease is an X-linked lysosomal storage disorder caused by mutations in the α-galactosidase A gene. Migalastat, a pharmacological chaperone, binds to specific mutant forms of α-galactosidase A to restore lysosomal activity.
Methods: A pharmacogenetic assay was used to identify the α-galactosidase A mutant forms amenable to migalastat. Six hundred Fabry disease-causing mutations were expressed in HEK-293 (HEK) cells; increases in α-galactosidase A activity were measured by a good laboratory practice (GLP)-validated assay (GLP HEK/Migalastat Amenability Assay). The predictive value of the assay was assessed based on pharmacodynamic responses to migalastat in phase II and III clinical studies.
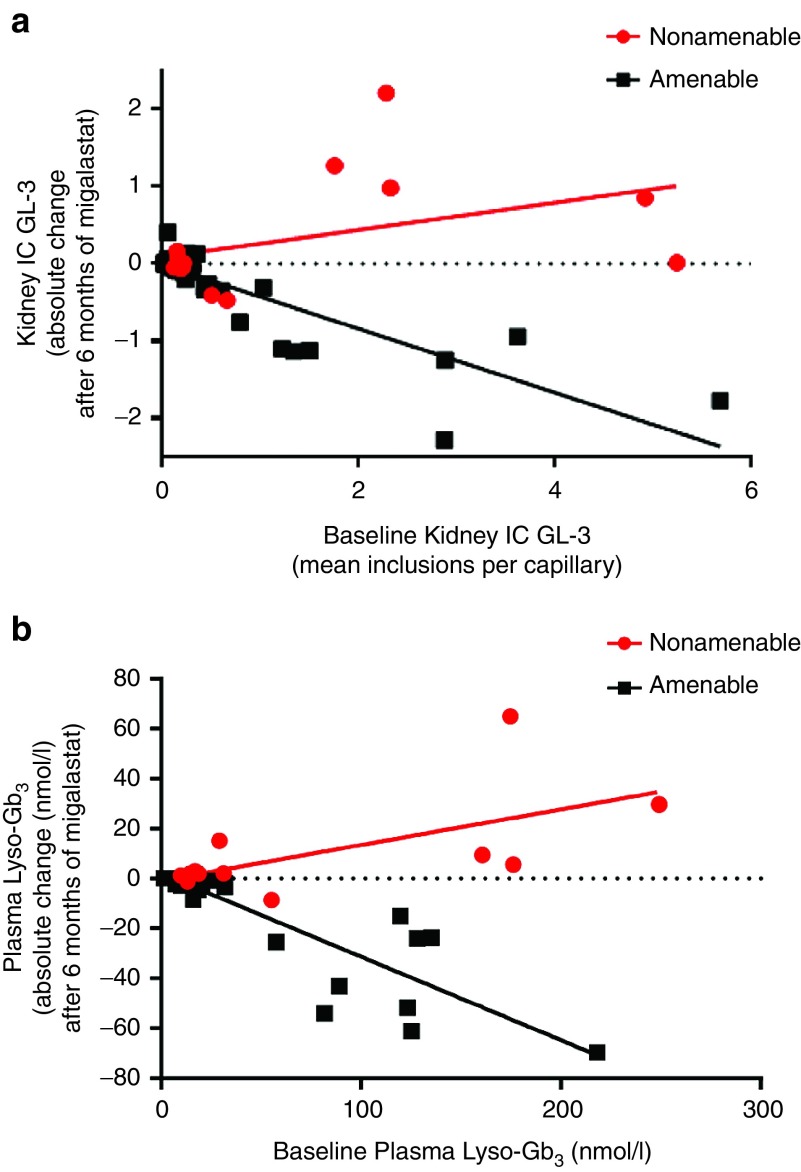
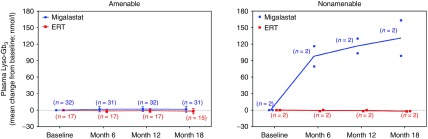
Results: Comparison of the GLP HEK assay results in in vivo white blood cell α-galactosidase A responses to migalastat in male patients showed high sensitivity, specificity, and positive and negative predictive values (≥0.875). GLP HEK assay results were also predictive of decreases in kidney globotriaosylceramide in males and plasma globotriaosylsphingosine in males and females. The clinical study subset of amenable mutations (n = 51) was representative of all 268 amenable mutations identified by the GLP HEK assay.
Conclusion: The GLP HEK assay is a clinically validated method of identifying male and female Fabry patients for treatment with migalastat.Genet Med 19 4, 430-438.
Trial registration: ClinicalTrials.gov NCT00214500 NCT00283959 NCT00283933 NCT00304512 NCT00925301 NCT01218659.
Figures
References
-
- Brady RO, Gal AE, Bradley RM, Martensson E, Warshaw AL, Laster L. Enzymatic defect in Fabry's disease. Ceramidetrihexosidase deficiency. N Engl J Med 1967;276:1163–1167. - PubMed
-
- Gal A, Schäfer E, Rohard I. The genetic basis of Fabry disease. In: Mehta A, Beck M, Sunder-Plassmann G (eds). Fabry Disease: Perspectives From 5 Years of FOS. Oxford PharmaGenesis: Oxford, UK, 2006. - PubMed
MeSH terms
Substances
Associated data
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
