

A prospective study on the natural history of patients with profound combined immunodeficiency: An interim analysis
- PMID: 27658761
- PMCID: PMC6311415
- DOI: 10.1016/j.jaci.2016.07.040
A prospective study on the natural history of patients with profound combined immunodeficiency: An interim analysis
Abstract
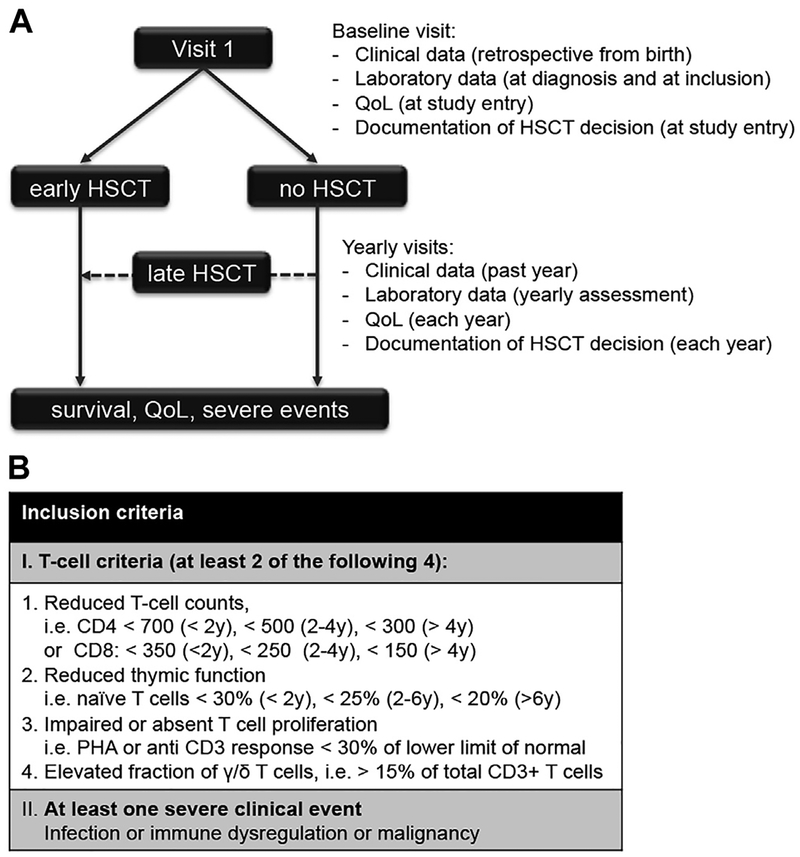
Background: Absent T-cell immunity resulting in life-threatening infections provides a clear rationale for hematopoetic stem cell transplantation (HSCT) in patients with severe combined immunodeficiency (SCID). Combined immunodeficiencies (CIDs) and "atypical" SCID show reduced, not absent T-cell immunity. If associated with infections or autoimmunity, they represent profound combined immunodeficiency (P-CID), for which outcome data are insufficient for unambiguous early transplant decisions.
Objectives: We sought to compare natural histories of severity-matched patients with/without subsequent transplantation and to determine whether immunologic and/or clinical parameters may be predictive for outcome.
Methods: In this prospective and retrospective observational study, we recruited nontransplanted patients with P-CID aged 1 to 16 years to compare natural histories of severity-matched patients with/without subsequent transplantation and to determine whether immunologic and/or clinical parameters may be predictive for outcome.
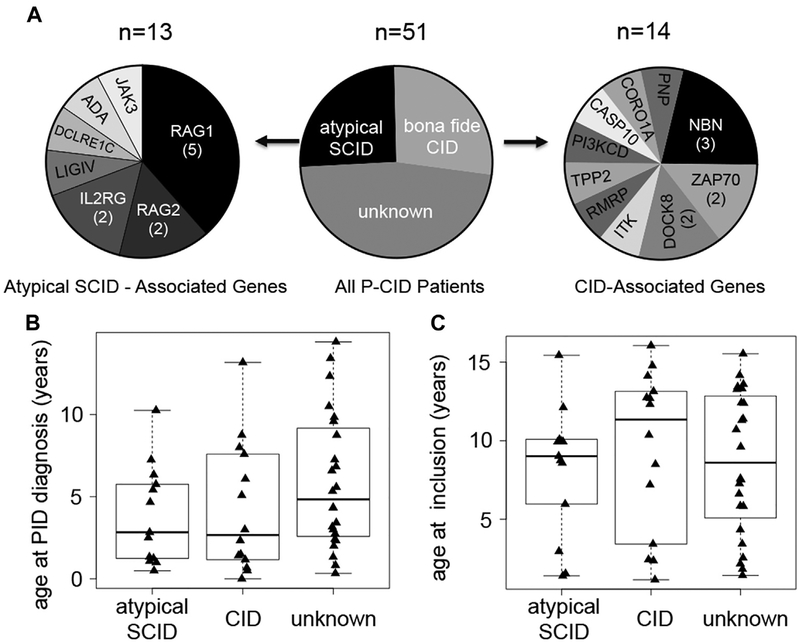
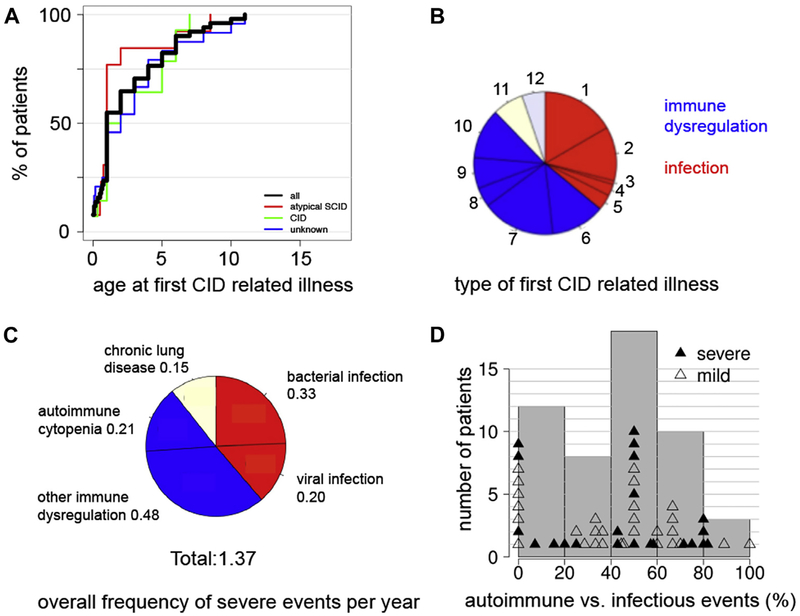
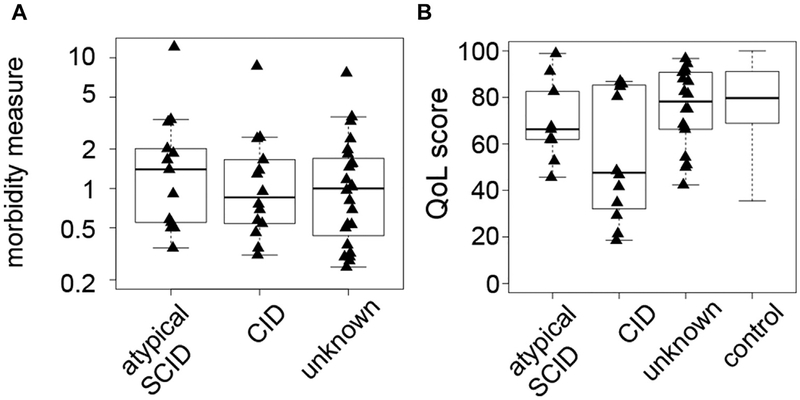
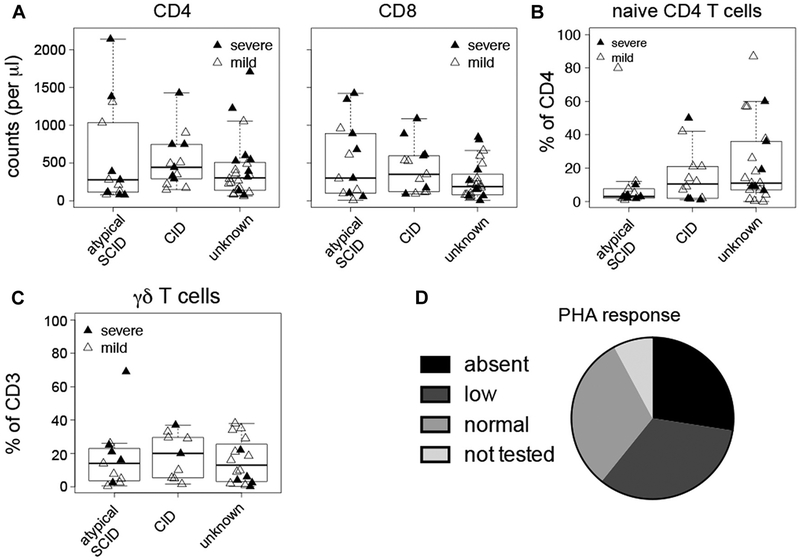
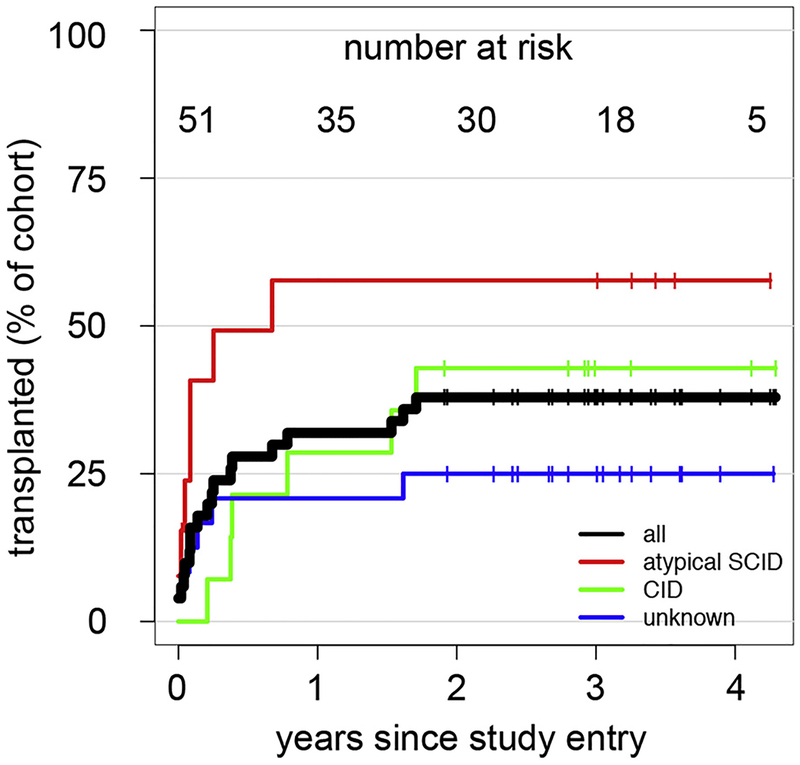
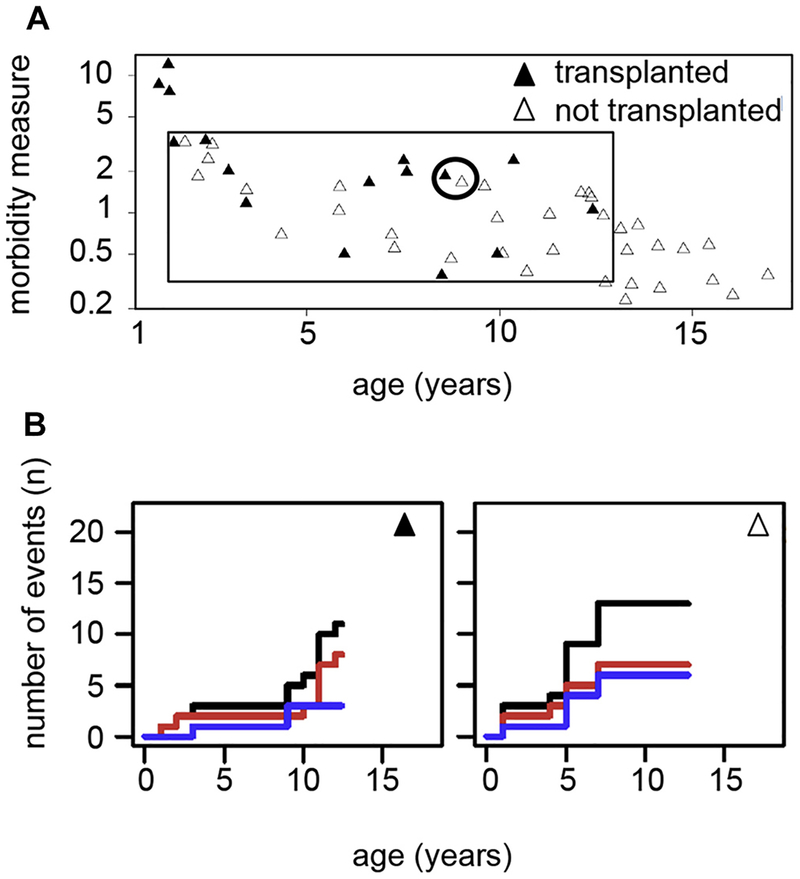
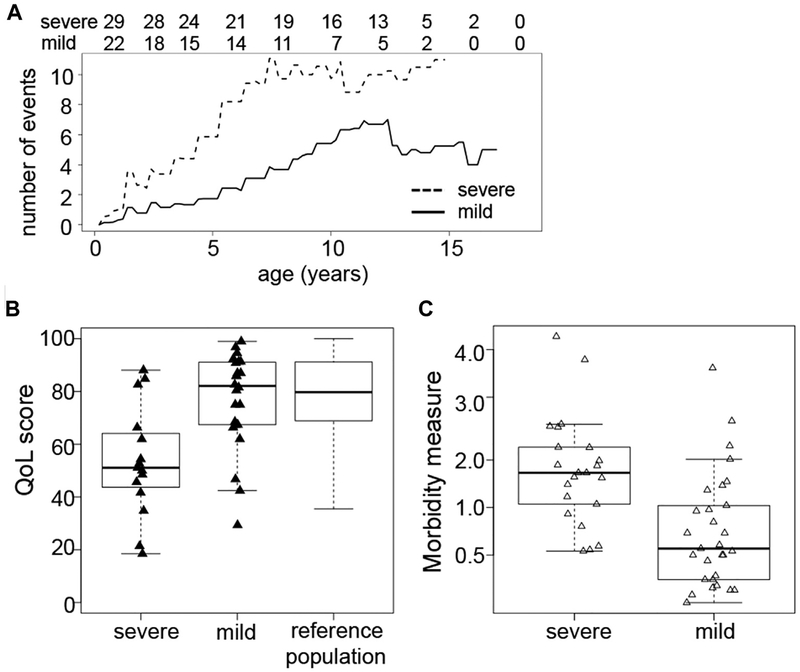
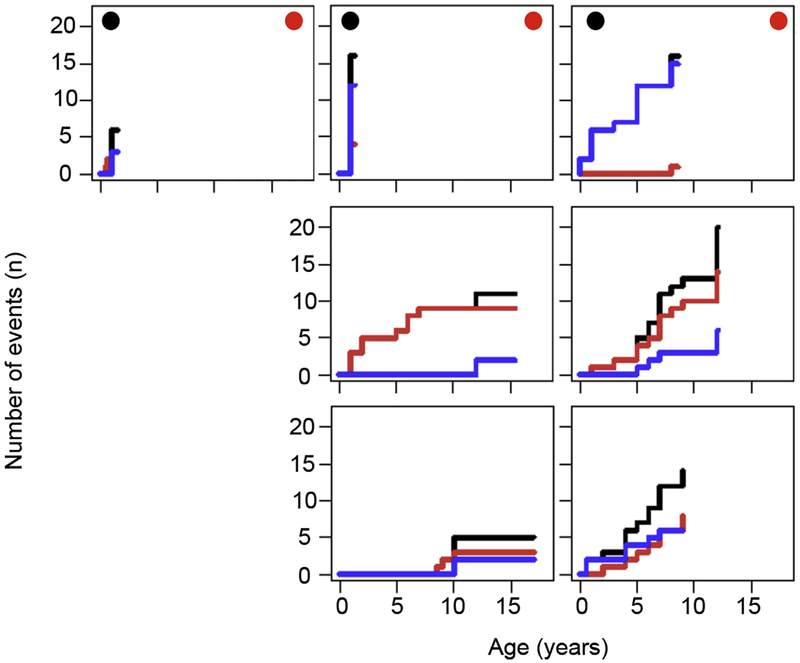
Results: A total of 51 patients were recruited (median age, 9.6 years). Thirteen of 51 had a genetic diagnosis of "atypical" SCID and 14 of 51 of CID. About half of the patients had less than 10% naive T cells, reduced/absent T-cell proliferation, and at least 1 significant clinical event/year, demonstrating their profound immunodeficiency. Nineteen patients (37%) underwent transplantation within 1 year of enrolment, and 5 of 51 patients died. Analysis of the HSCT decisions revealed the anticipated heterogeneity, favoring an ongoing prospective matched-pair analysis of patients with similar disease severity with or without transplantation. Importantly, so far neither the genetic diagnosis nor basic measurements of T-cell immunity were good predictors of disease evolution.
Conclusions: The P-CID study for the first time characterizes a group of patients with nontypical SCID T-cell deficiencies from a therapeutic perspective. Because genetic and basic T-cell parameters provide limited guidance, prospective data from this study will be a helpful resource for guiding the difficult HSCT decisions in patients with P-CID.
Keywords: T-cell deficiency; combined immunodeficiency; hematopoietic stem cell transplantation; natural history.
Copyright © 2016 American Academy of Allergy, Asthma & Immunology. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Disclosure of potential conflict of interest: C. Speckmann receives grant support from the German Ministry for Education and Research (BMBF); payments for lectures from Octapharma and CSL Behring; and travel support from Orphan Europe and CSL Behring. A. Aiuti receives grant support from the European Research Council, Fondazione Telethon Rome, Italian Ministry of Health, and GSK. M. H. Albert receives grant support from GSK; receives payment for lectures from Biotest, MSD, and Jazz; holds stock in Amen and BMS; and receives travel support from Octapharma. T. Avcin serves as a consultant for Octapharma and receives payment for lectures from Octapharma. C. Cancrini receives grant support from Ministero della Salute and European Community and serves as a consultant for UCB CellTech UK. A. Finocchi receives grant support from Telethon. H. Bobby Gaspar serves as a consultant for Orchard Therapeutics and holds stock in Orchard Therapeutis. K. Gilmour receives travel support from UKPIN. M. Hoenig receives payment for lectures from CSL Behring and Jazz Pharma and travel support from CSL Behring and Jazz Pharma. L. Notarangelo serves on the board of Novimmune and receives grant support from the National Institutes of Health and royalties from Up-to-Date. M. G. Seidel serves as a consultant for Baxalta and Novartis; received payments for lectures from Jazz Pharmaceuticals, Novartis, and CSL Behring; and received travel support from Jazz Pharmaceuticals, Octapharma, and Amgen. P. Soler-Palacin receives travel support from the P-CID study group; expert testimony from CSL Behring, Octapharma, Grifols, and Baxter; grant support from CSL Behring; and payments for lectures from Grifols. K. Warnatz serves on the board for BioTest, CSL Behring, and LFB Biomedicaments; receives grant support from BMS, CSL Behring, and BioTest; and receives payments for lecture from LFB Biomedicaments, Baxter, GSK, CSL Behring, Pfizer, BioTest, Novartis Pharma, Roche, Octapharma, and UCB Pharma. A. Worth receives research support from the National Institute of Health Research and Wellcome Trust. A. Uhlmann receives travel support from BMBF. S. Ehl receives research support from BMBF, the Canadian Immunodeficiency Society, and European Union’s Horizon 2020 Research and Innovation Programme; serves as a consultant for UCD and Novartis but not in the context of this study; and received payments from lectures for CSL Behring. The rest of the authors declare that they have no relevant conflicts of interest.
Figures
References
-
- Buckley R Primary immunodeficiency diseases due to defects in lymphocytes. N Engl J Med 2000;343:1313–24. - PubMed
-
- Notarangelo LD. Primary immunodeficiencies. J Allergy Clin Immunol 2010;125: S182–94. - PubMed
-
- Roifman CM, Somech R, Kavadas F, Pires L, Nahum A, Dalal I, et al. Defining combined immunodeficiency. J Allergy Clin Immunol 2012;130:177–83. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
