

Structured States of Disordered Proteins from Genomic Sequences
- PMID: 27662088
- PMCID: PMC5451116
- DOI: 10.1016/j.cell.2016.09.010
Structured States of Disordered Proteins from Genomic Sequences
Abstract
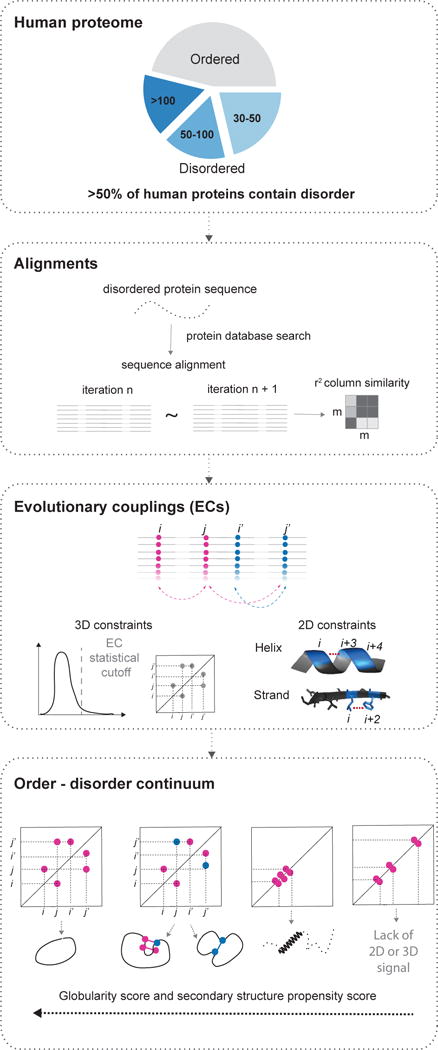
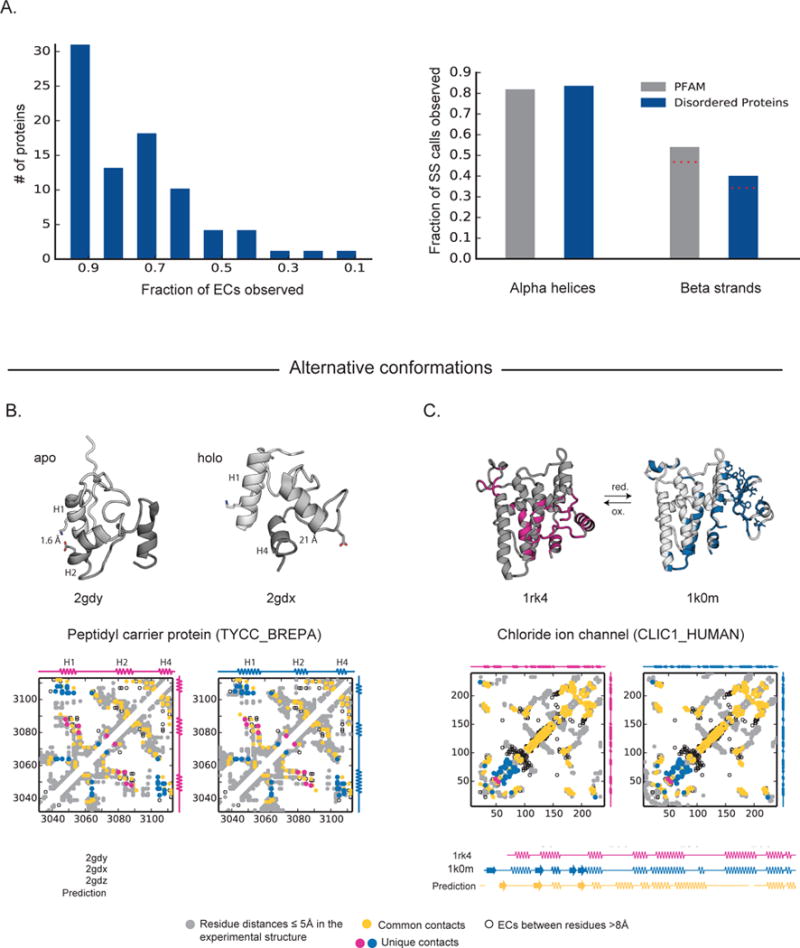
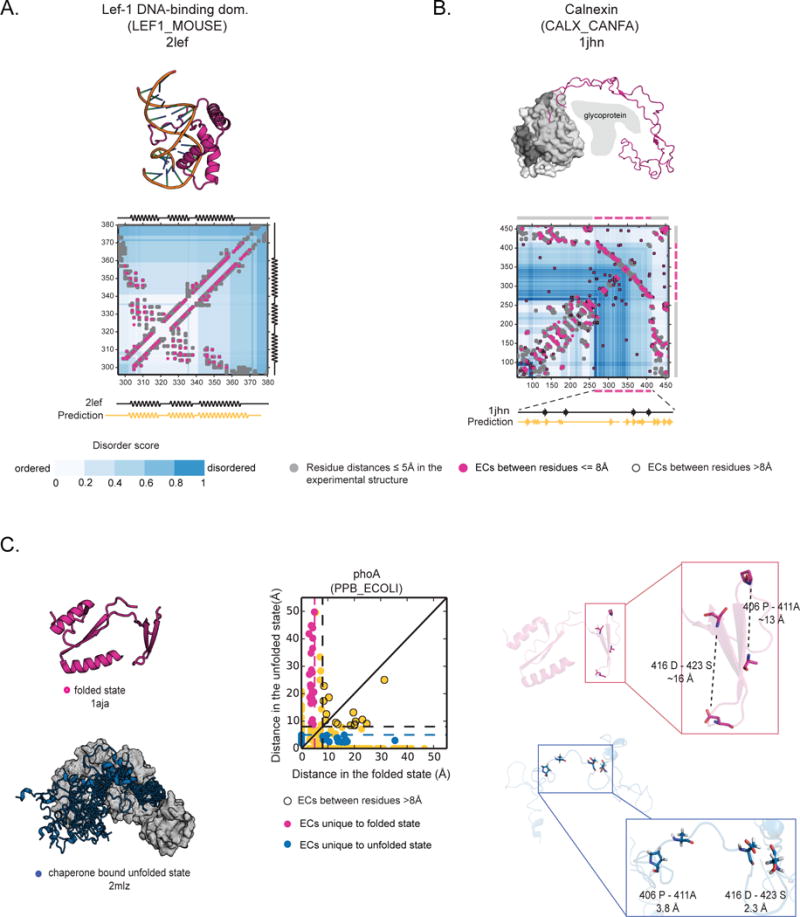
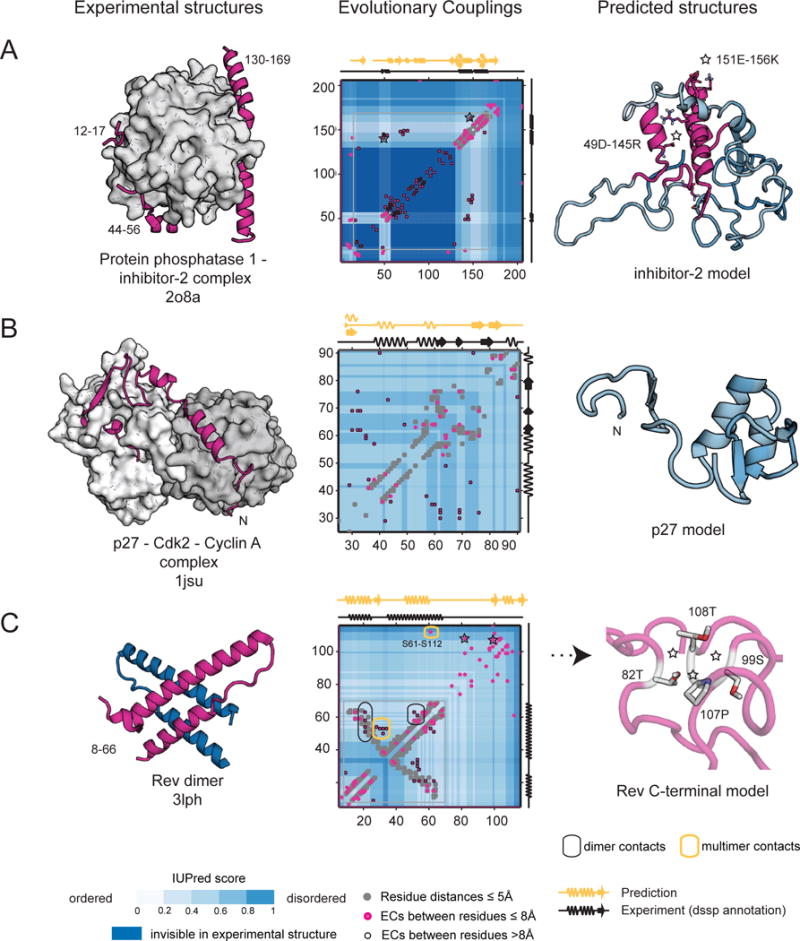
Protein flexibility ranges from simple hinge movements to functional disorder. Around half of all human proteins contain apparently disordered regions with little 3D or functional information, and many of these proteins are associated with disease. Building on the evolutionary couplings approach previously successful in predicting 3D states of ordered proteins and RNA, we developed a method to predict the potential for ordered states for all apparently disordered proteins with sufficiently rich evolutionary information. The approach is highly accurate (79%) for residue interactions as tested in more than 60 known disordered regions captured in a bound or specific condition. Assessing the potential for structure of more than 1,000 apparently disordered regions of human proteins reveals a continuum of structural order with at least 50% with clear propensity for three- or two-dimensional states. Co-evolutionary constraints reveal hitherto unseen structures of functional importance in apparently disordered proteins.
Keywords: EVfold; Evolutionary couplings; bioinformatics; computational biology; conformational flexibility; disorder; maximum entropy; statistical physics; structure prediction.
Copyright © 2016 Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
-
- Bah A, Vernon RM, Siddiqui Z, Krzeminski M, Muhandiram R, Zhao C, Sonenberg N, Kay LE, Forman-Kay JD. Folding of an intrinsically disordered protein by phosphorylation as a regulatory switch. Nature. 2015;519:106–109. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
