

Mitochondrial Dysfunction and Synaptic Transmission Failure in Alzheimer's Disease
- PMID: 27662318
- PMCID: PMC5605817
- DOI: 10.3233/JAD-160702
Mitochondrial Dysfunction and Synaptic Transmission Failure in Alzheimer's Disease
Abstract
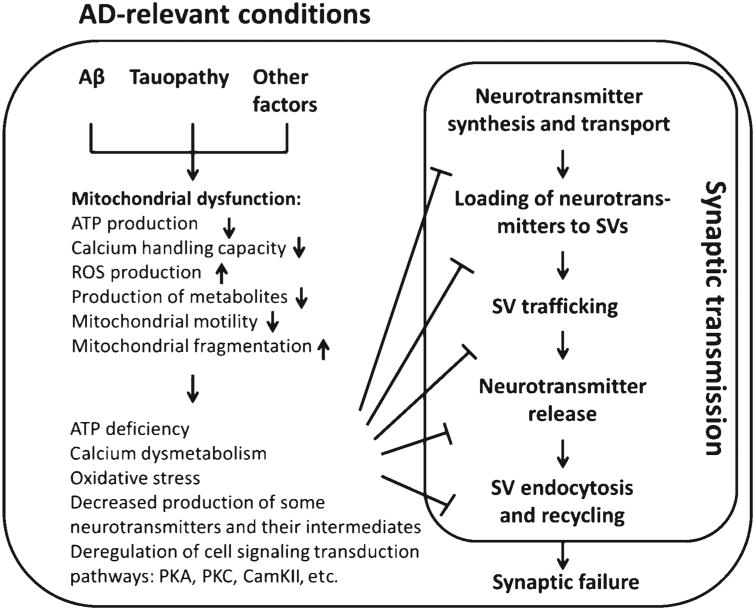
Alzheimer's disease (AD) is a chronic neurodegenerative disorder, in which multiple risk factors converge. Despite the complexity of the etiology of the disease, synaptic failure is the pathological basis of cognitive impairment, the cardinal sign of AD. Decreased synaptic density, compromised synaptic transmission, and defected synaptic plasticity are hallmark synaptic pathologies accompanying AD. However, the mechanisms by which synapses are injured in AD-related conditions have not been fully elucidated. Mitochondria are a critical organelle in neurons. The pivotal role of mitochondria in supporting synaptic function and the concomitant occurrence of mitochondrial dysfunction with synaptic stress in postmortem AD brains as well as AD animal models seem to lend the credibility to the hypothesis that mitochondrial defects underlie synaptic failure in AD. This concept is further strengthened by the protective effect of mitochondrial medicine on synaptic function against the toxicity of amyloid-β, a key player in the pathogenesis of AD. In this review, we focus on the association between mitochondrial dysfunction and synaptic transmission deficits in AD. Impaired mitochondrial energy production, deregulated mitochondrial calcium handling, excess mitochondrial reactive oxygen species generation and release play a crucial role in mediating synaptic transmission deregulation in AD. The understanding of the role of mitochondrial dysfunction in synaptic stress may lead to novel therapeutic strategies for the treatment of AD through the protection of synaptic transmission by targeting to mitochondrial deficits.
Keywords: Alzheimer’s disease; mitochondrial dysfunction; synaptic injury; synaptic mitochondria; synaptic transmission.
Figures
References
-
- Querfurth HW, LaFerla FM. Alzheimer's disease. N Engl J Med. 2010;362:329–344. - PubMed
-
- Kidd PM. Alzheimer's disease, amnestic mild cognitive impairment, and age-associated memory impairment: Current understanding and progress toward integrative prevention. Altern Med Rev. 2008;13:85–115. - PubMed
-
- Blennow K, de Leon MJ, Zetterberg H. Alzheimer's disease. Lancet. 2006;368:387–403. - PubMed
-
- Landon M, Kidd M. Amyloid in Alzheimer's disease. Biochem Soc Trans. 1989;17:69–72. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
