

The chromatin-binding protein Smyd1 restricts adult mammalian heart growth
- PMID: 27663768
- PMCID: PMC5130490
- DOI: 10.1152/ajpheart.00235.2016
The chromatin-binding protein Smyd1 restricts adult mammalian heart growth
Abstract
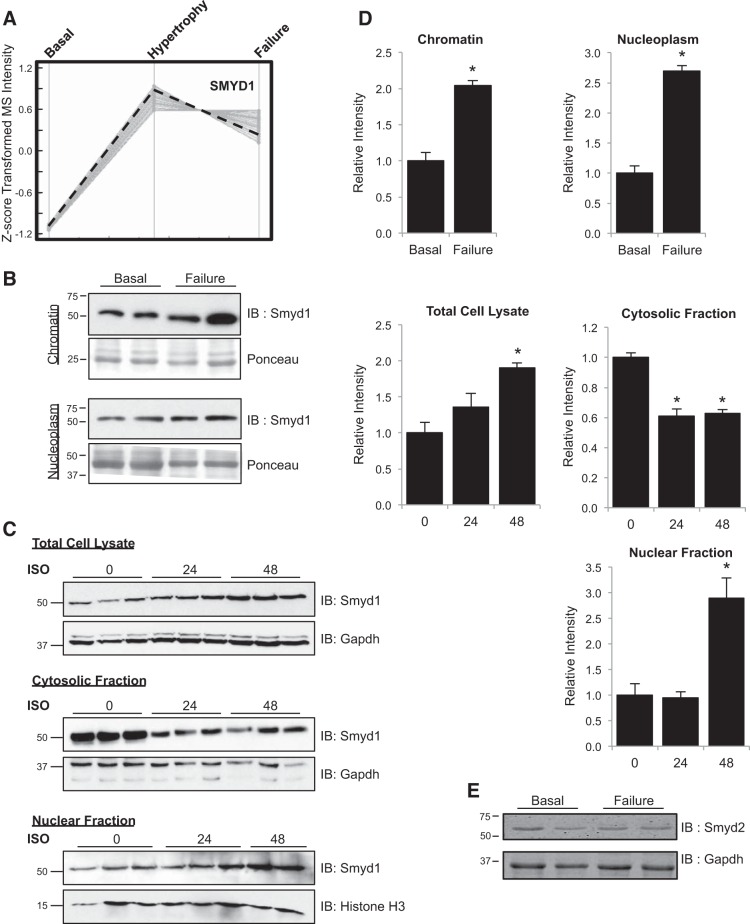
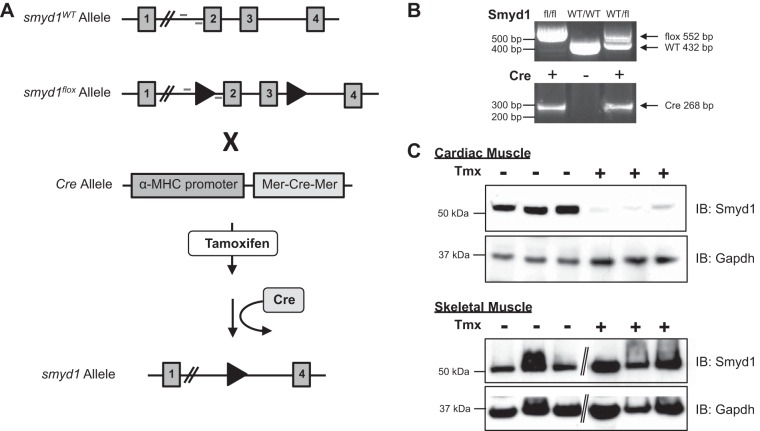
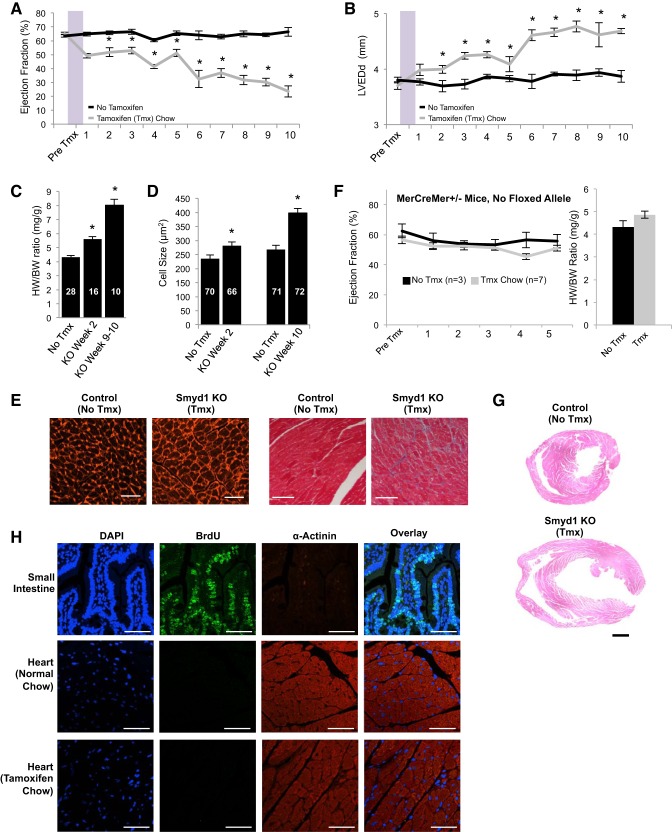
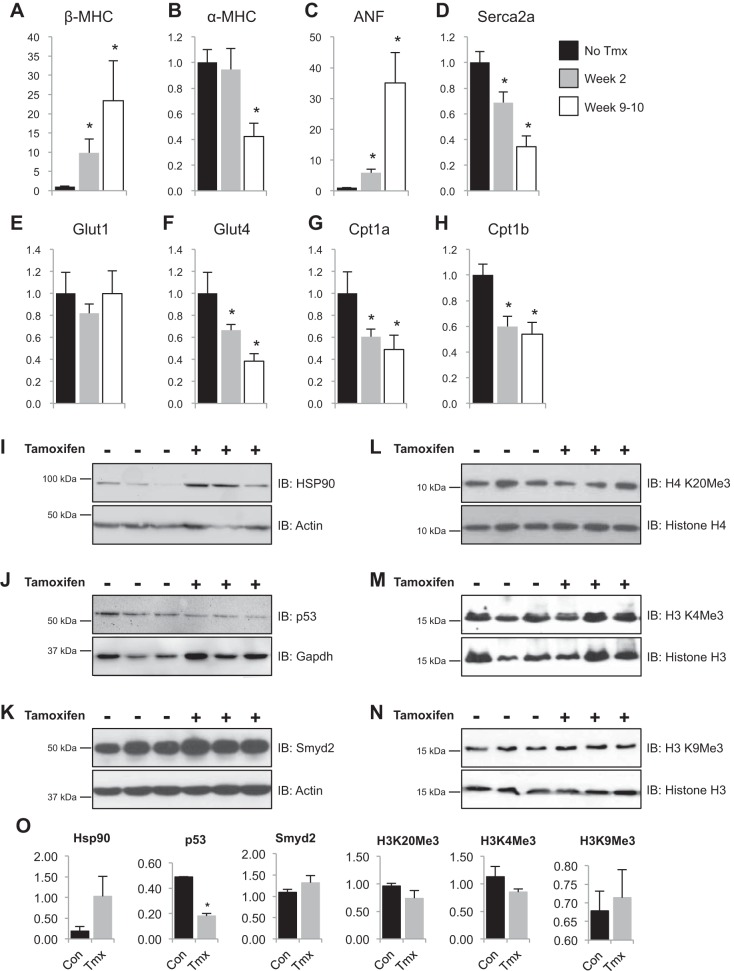
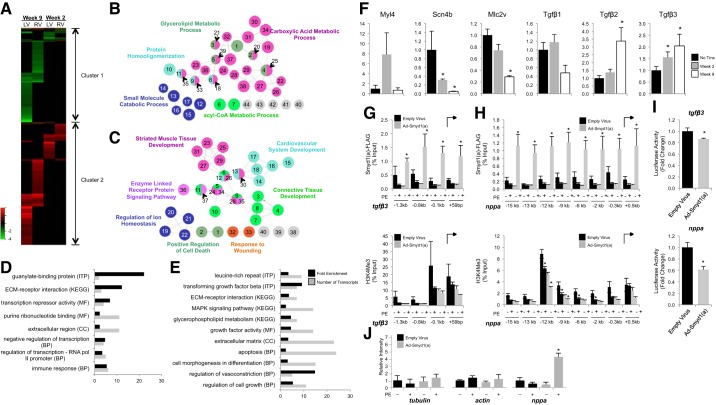
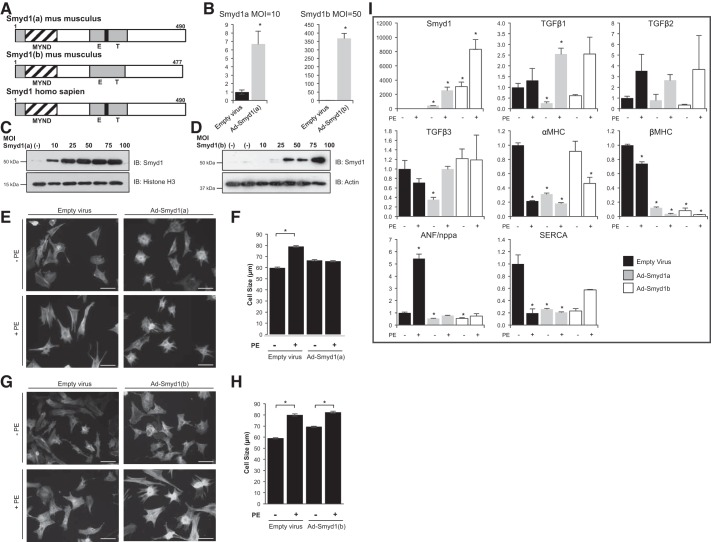
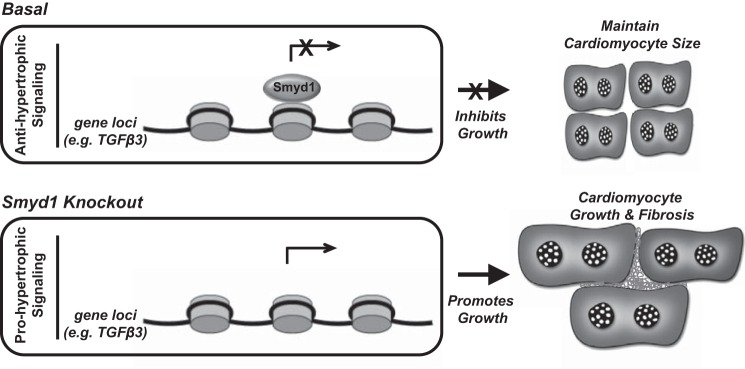
All terminally differentiated organs face two challenges, maintaining their cellular identity and restricting organ size. The molecular mechanisms responsible for these decisions are of critical importance to organismal development, and perturbations in their normal balance can lead to disease. A hallmark of heart failure, a condition affecting millions of people worldwide, is hypertrophic growth of cardiomyocytes. The various forms of heart failure in human and animal models share conserved transcriptome remodeling events that lead to expression of genes normally silenced in the healthy adult heart. However, the chromatin remodeling events that maintain cell and organ size are incompletely understood; insights into these mechanisms could provide new targets for heart failure therapy. Using a quantitative proteomics approach to identify muscle-specific chromatin regulators in a mouse model of hypertrophy and heart failure, we identified upregulation of the histone methyltransferase Smyd1 during disease. Inducible loss-of-function studies in vivo demonstrate that Smyd1 is responsible for restricting growth in the adult heart, with its absence leading to cellular hypertrophy, organ remodeling, and fulminate heart failure. Molecular studies reveal Smyd1 to be a muscle-specific regulator of gene expression and indicate that Smyd1 modulates expression of gene isoforms whose expression is associated with cardiac pathology. Importantly, activation of Smyd1 can prevent pathological cell growth. These findings have basic implications for our understanding of cardiac pathologies and open new avenues to the treatment of cardiac hypertrophy and failure by modulating Smyd1.
Keywords: Smyd1; cardiac hypertrophy; epigenetics; heart failure; histone methyltransferase.
Copyright © 2016 the American Physiological Society.
Figures
References
-
- Abaci N, Gulec C, Bayrak F, Komurcu Bayrak E, Kahveci G, Erginel Unaltuna N. The variations of BOP gene in hypertrophic cardiomyopathy. Anadolu Kardiyol Derg 10: 303–309, 2010. - PubMed
-
- Abu-Farha M, Lambert JP, Al-Madhoun AS, Elisma F, Skerjanc IS, Figeys D. The tale of two domains: Proteomics and genomics analysis of SMYD2, a new histone methyltransferase. Mol Cell Proteomics 7: 560–572, 2008. - PubMed
-
- Ago T, Liu T, Zhai P, Chen W, Li H, Molkentin JD, Vatner SF, Sadoshima J. A redox-dependent pathway for regulating class II HDACs and cardiac hypertrophy. Cell 133: 978–993, 2008. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases