

Guidelines for Large-Scale Sequence-Based Complex Trait Association Studies: Lessons Learned from the NHLBI Exome Sequencing Project
- PMID: 27666372
- PMCID: PMC5065683
- DOI: 10.1016/j.ajhg.2016.08.012
Guidelines for Large-Scale Sequence-Based Complex Trait Association Studies: Lessons Learned from the NHLBI Exome Sequencing Project
Abstract
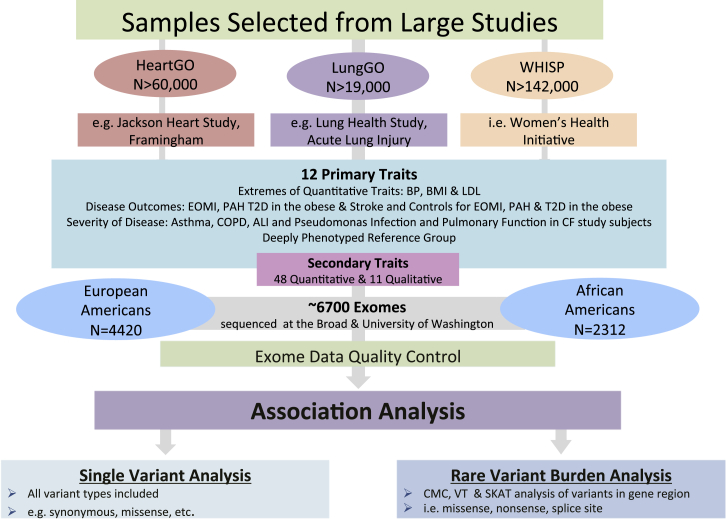
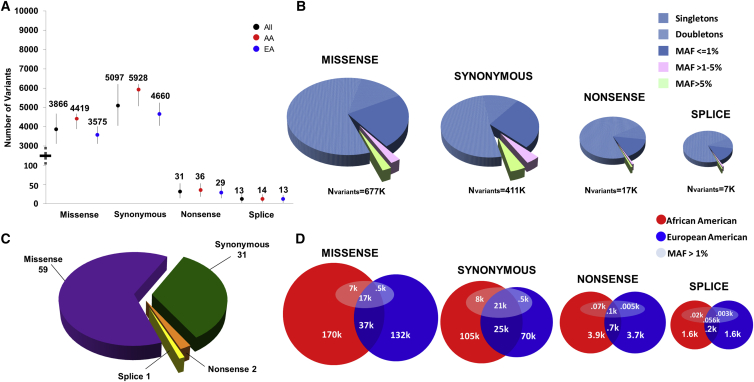
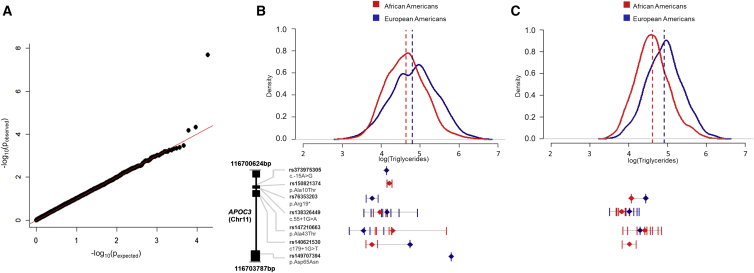
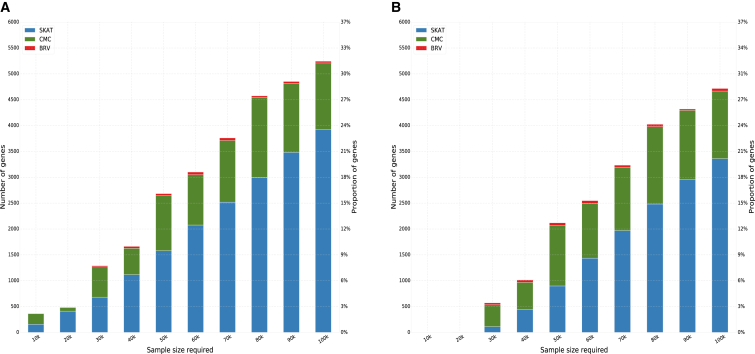
Massively parallel whole-genome sequencing (WGS) data have ushered in a new era in human genetics. These data are now being used to understand the role of rare variants in complex traits and to advance the goals of precision medicine. The technological and computing advances that have enabled us to generate WGS data on thousands of individuals have also outpaced our ability to perform analyses in scientifically and statistically rigorous and thoughtful ways. The past several years have witnessed the application of whole-exome sequencing (WES) to complex traits and diseases. From our analysis of NHLBI Exome Sequencing Project (ESP) data, not only have a number of important disease and complex trait association findings emerged, but our collective experience offers some valuable lessons for WGS initiatives. These include caveats associated with generating automated pipelines for quality control and analysis of rare variants; the importance of studying minority populations; sample size requirements and efficient study designs for identifying rare-variant associations; and the significance of incidental findings in population-based genetic research. With the ESP as an example, we offer guidance and a framework on how to conduct a large-scale association study in the era of WGS.
Copyright © 2016 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
