

Smoothing a rugged protein folding landscape by sequence-based redesign
- PMID: 27667094
- PMCID: PMC5036219
- DOI: 10.1038/srep33958
Smoothing a rugged protein folding landscape by sequence-based redesign
Abstract
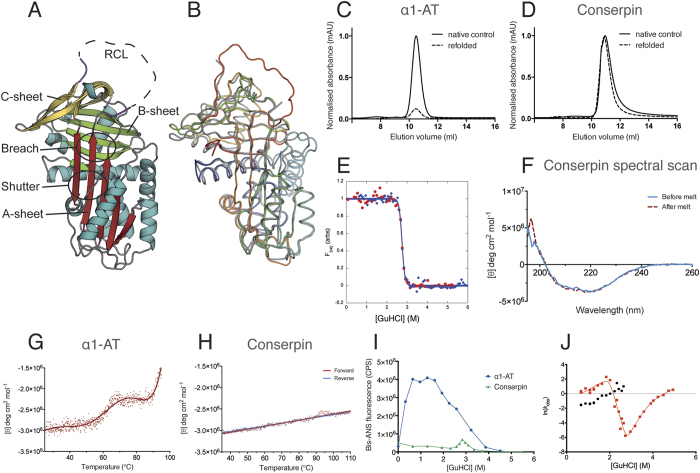
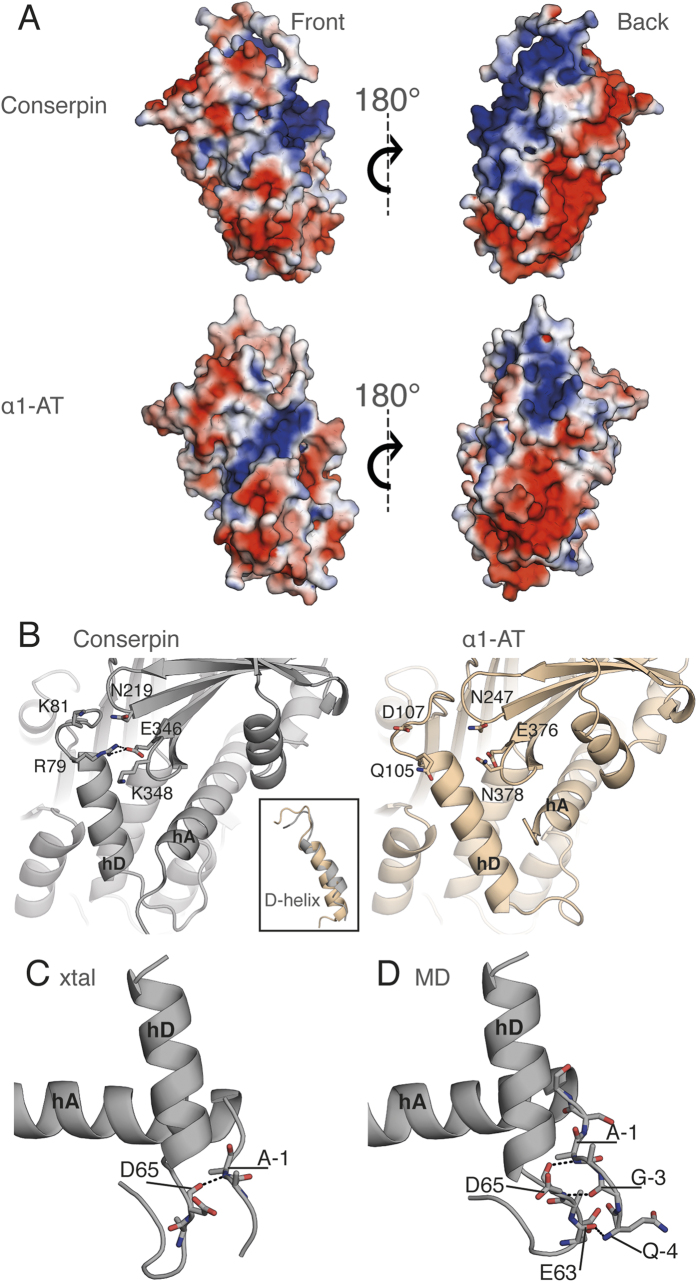
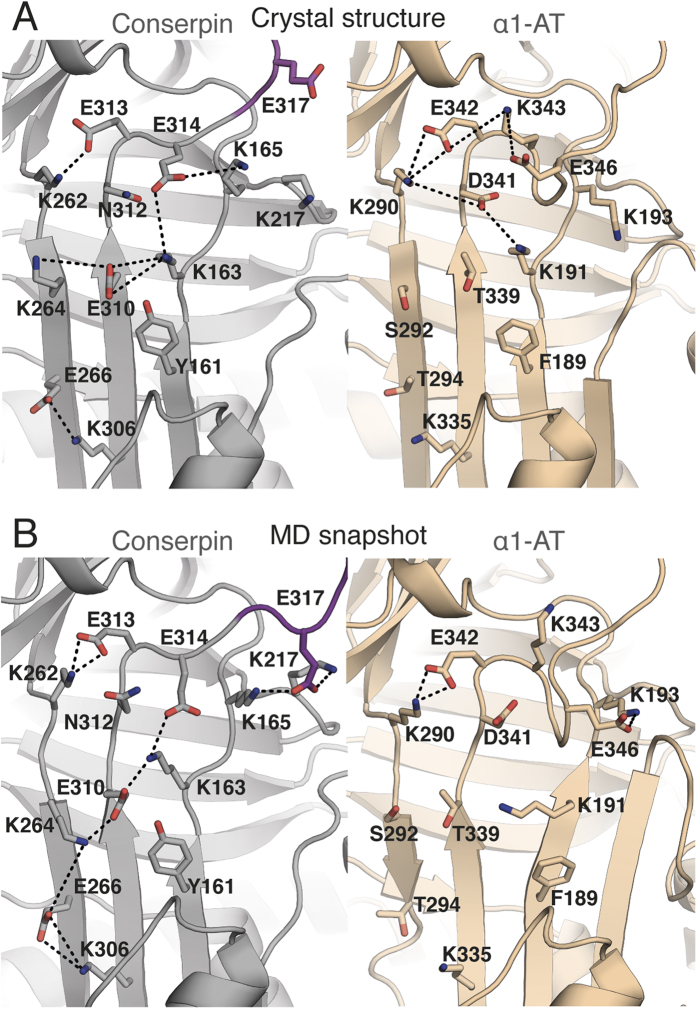
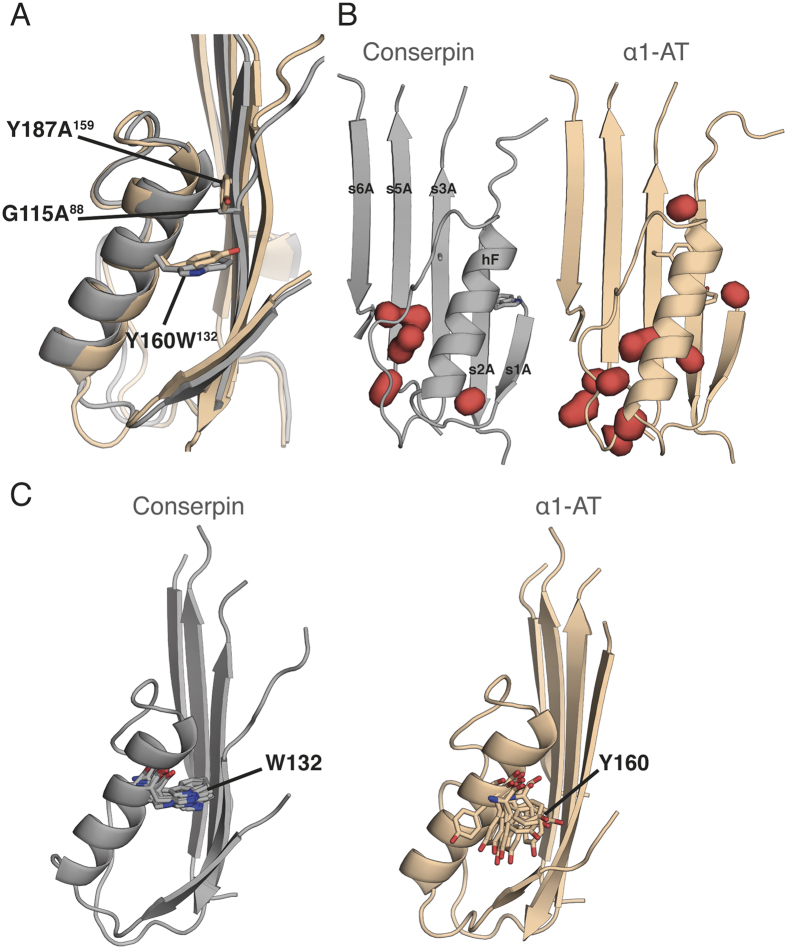
The rugged folding landscapes of functional proteins puts them at risk of misfolding and aggregation. Serine protease inhibitors, or serpins, are paradigms for this delicate balance between function and misfolding. Serpins exist in a metastable state that undergoes a major conformational change in order to inhibit proteases. However, conformational labiality of the native serpin fold renders them susceptible to misfolding, which underlies misfolding diseases such as α1-antitrypsin deficiency. To investigate how serpins balance function and folding, we used consensus design to create conserpin, a synthetic serpin that folds reversibly, is functional, thermostable, and polymerization resistant. Characterization of its structure, folding and dynamics suggest that consensus design has remodeled the folding landscape to reconcile competing requirements for stability and function. This approach may offer general benefits for engineering functional proteins that have risky folding landscapes, including the removal of aggregation-prone intermediates, and modifying scaffolds for use as protein therapeutics.
Figures
Similar articles
-
Reactive centre loop dynamics and serpin specificity.Sci Rep. 2019 Mar 7;9(1):3870. doi: 10.1038/s41598-019-40432-w. Sci Rep. 2019. PMID: 30846766 Free PMC article.
-
Probing the folding pathway of a consensus serpin using single tryptophan mutants.Sci Rep. 2018 Feb 1;8(1):2121. doi: 10.1038/s41598-018-19567-9. Sci Rep. 2018. PMID: 29391487 Free PMC article.
-
Crystallography of serpins and serpin complexes.Methods Enzymol. 2011;501:63-87. doi: 10.1016/B978-0-12-385950-1.00005-5. Methods Enzymol. 2011. PMID: 22078531
-
Molecular gymnastics: serpin structure, folding and misfolding.Curr Opin Struct Biol. 2006 Dec;16(6):761-8. doi: 10.1016/j.sbi.2006.10.005. Epub 2006 Oct 31. Curr Opin Struct Biol. 2006. PMID: 17079131 Review.
-
The native metastability and misfolding of serine protease inhibitors.Protein Pept Lett. 2005 Jul;12(5):477-81. doi: 10.2174/0929866054395365. Protein Pept Lett. 2005. PMID: 16029161 Review.
Cited by
-
The stability and dynamics of computationally designed proteins.Protein Eng Des Sel. 2022 Feb 17;35:gzac001. doi: 10.1093/protein/gzac001. Protein Eng Des Sel. 2022. PMID: 35174855 Free PMC article. Review.
-
Engineering the serpin α1 -antitrypsin: A diversity of goals and techniques.Protein Sci. 2020 Apr;29(4):856-871. doi: 10.1002/pro.3794. Epub 2019 Dec 9. Protein Sci. 2020. PMID: 31774589 Free PMC article. Review.
-
Comparison of structures and inhibition activities of serine protease inhibitors of Trichinella spiralis and Trichinella pseudospiralis.Cell Biosci. 2025 Mar 13;15(1):35. doi: 10.1186/s13578-025-01375-0. Cell Biosci. 2025. PMID: 40082967 Free PMC article.
-
Protein stability is determined by single-site bias rather than pairwise covariance.bioRxiv [Preprint]. 2025 Jan 14:2025.01.09.632118. doi: 10.1101/2025.01.09.632118. bioRxiv. 2025. PMID: 39868188 Free PMC article. Preprint.
-
Consensus sequence design as a general strategy to create hyperstable, biologically active proteins.Proc Natl Acad Sci U S A. 2019 Jun 4;116(23):11275-11284. doi: 10.1073/pnas.1816707116. Epub 2019 May 20. Proc Natl Acad Sci U S A. 2019. PMID: 31110018 Free PMC article.
References
-
- Dill K. A. & Chan H. S. From Levinthal to pathways to funnels. Nat. Struct. Biol. 4, 10–19 (1997). - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
