

Episodic Ataxias: Clinical and Genetic Features
- PMID: 27667184
- PMCID: PMC5035943
- DOI: 10.14802/jmd.16028
Episodic Ataxias: Clinical and Genetic Features
Abstract
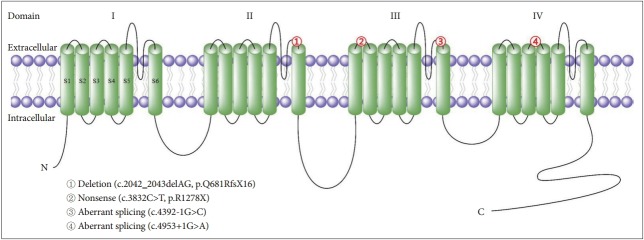
Episodic ataxia (EA) is a clinically heterogeneous group of disorders that are characterized by recurrent spells of truncal ataxia and incoordination lasting minutes to hours. Most have an autosomal dominant inheritance pattern. To date, 8 subtypes have been defined according to clinical and genetic characteristics, and five genes are known to be linked to EAs. Both EA1 and EA2, which are caused by mutations in KCNA1 and CACNA1A, account for the majority of EA, but many patients with no identified mutations still exhibit EA-like clinical features. Furthermore, genetically confirmed EAs have mostly been identified in Caucasian families. In this article, we review the current knowledge on the clinical and genetic characteristics of EAs. Additionally, we summarize the phenotypic features of the genetically confirmed EA2 families in Korea.
Keywords: CACNA1A; Episodic ataxia; KCNA1.
Conflict of interest statement
The authors have no financial conflicts of interest.
Figures
References
-
- Jen JC, Graves TD, Hess EJ, Hanna MG, Griggs RC, Baloh RW, CINCH investigators Primary episodic ataxias: diagnosis, pathogenesis and treatment. Brain. 2007;130(Pt 10):2484–2493. - PubMed
-
- Tomlinson SE, Hanna MG, Kullmann DM, Tan SV, Burke D. Clinical neurophysiology of the episodic ataxias: insights into ion channel dysfunction in vivo. Clin Neurophysiol. 2009;120:1768–1776. - PubMed
-
- Jen JC, Wan J, Palos TP, Howard BD, Baloh RW. Mutation in the glutamate transporter EAAT1 causes episodic ataxia, hemiplegia, and seizures. Neurology. 2005;65:529–534. - PubMed
-
- de Vries B, Mamsa H, Stam AH, Wan J, Bakker SL, Vanmolkot KR, et al. Episodic ataxia associated with EAAT1 mutation C186S affecting glutamate reuptake. Arch Neurol. 2009;66:97–101. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
