

Clinical and genetic aspects of KBG syndrome
- PMID: 27667800
- PMCID: PMC5435101
- DOI: 10.1002/ajmg.a.37842
Clinical and genetic aspects of KBG syndrome
Abstract
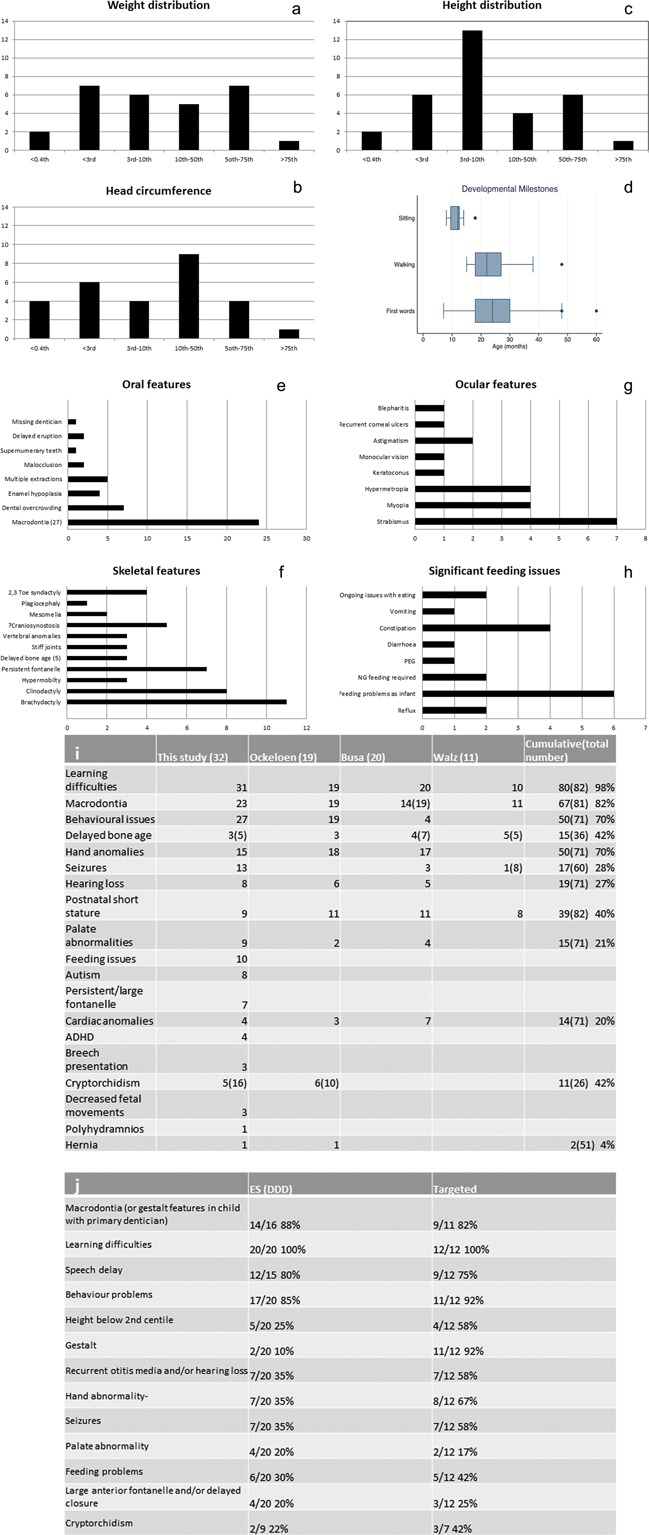
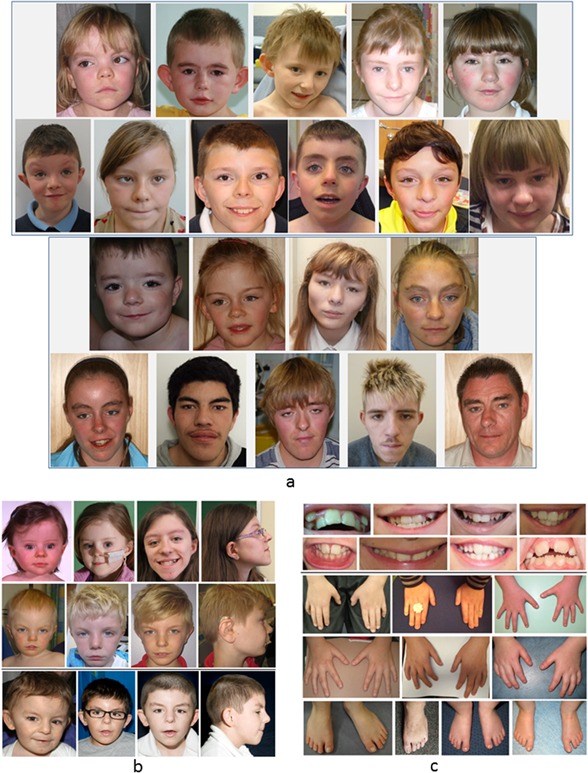
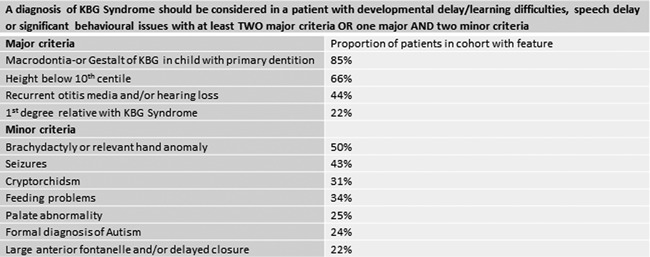
KBG syndrome is characterized by short stature, distinctive facial features, and developmental/cognitive delay and is caused by mutations in ANKRD11, one of the ankyrin repeat-containing cofactors. We describe 32 KBG patients aged 2-47 years from 27 families ascertained via two pathways: targeted ANKRD11 sequencing (TS) in a group who had a clinical diagnosis of KBG and whole exome sequencing (ES) in a second group in whom the diagnosis was unknown. Speech delay and learning difficulties were almost universal and variable behavioral problems frequent. Macrodontia of permanent upper central incisors was seen in 85%. Other clinical features included short stature, conductive hearing loss, recurrent middle ear infection, palatal abnormalities, and feeding difficulties. We recognized a new feature of a wide anterior fontanelle with delayed closure in 22%. The subtle facial features of KBG syndrome were recognizable in half the patients. We identified 20 ANKRD11 mutations (18 novel: all truncating) confirmed by Sanger sequencing in 32 patients. Comparison of the two ascertainment groups demonstrated that facial/other typical features were more subtle in the ES group. There were no conclusive phenotype-genotype correlations. Our findings suggest that mutation of ANKRD11 is a common Mendelian cause of developmental delay. Affected patients may not show the characteristic KBG phenotype and the diagnosis is therefore easily missed. We propose updated diagnostic criteria/clinical recommendations for KBG syndrome and suggest that inclusion of ANKRD11 will increase the utility of gene panels designed to investigate developmental delay. © 2016 The Authors. American Journal of Medical Genetics Part A Published by Wiley Periodicals, Inc.
Keywords: ANKRD11; KBG syndrome; macrodontia.
© 2016 The Authors. American Journal of Medical Genetics Part A Published by Wiley Periodicals, Inc.
Figures
References
-
- Ansari M, Poke G, Ferry Q, Williamson K, Aldridge R, Meynert AM, Bengani H, Yee Chan C, Kayserili H, Avci S, Hennekam RCM, Lampe AK, Redeker E, Homfray T, Ross A, Falkenberg Smeland M, Mansour S, Parker MJ, Cook JA, Splitt M, Fisher RB, Fryer A, Magee AC, Wilkie A, Barnicoat A, Brady AF, Cooper NS, Mercer C, Deshpande C, Bennett CP, Pilz DT, Ruddy D, Cilliers D, Johnson DS, Josifova D, Rosser E, Thompson EM, Wakeling E, Kinning E, Stewart F, Flinter F, Girisha KM, Cox H, Firth HV, Kingstn H, Wee JS, Hurst JA, Clayton‐Smith J, Tolmie J, Vogt J, Tatton‐Brown K, Chandler K, Prescott K, Wilson L, Behnam M, McEntagart M, Davidson R, Lynch S, Sisodiya S, Mehta SG, McKee SA, Mohammed S, Holden S, Park S, Holder SE, Harrison V, McConnell V, Lam WK, Green AJ, Donnai D, Bitner‐Glindzicz M, Donnelly DE, Nellaker C, Taylor MS, Fitzpatrick DR. 2014. Genetic heterogeneity in Cornelia de Lange syndrome (CdLS) and CdLS‐like phenotypes with observed and predicted levels of mosaicism. J Med Genet 51:659–668. - PMC - PubMed
-
- Ashraf T, Irving M, Canham N, Holder S, Foulds N, Magee A, McConnell V, Fisher R, McEntagart M, Tolmie J, Joss S, Clayton‐Smith J, Study DDD. 2015. KBG syndrome: A DDD front‐runner? Eur Soc Hum Genet Abstract PM08 38 159.
-
- Busa T, Tessier A, Riccardi F, Jacquette A, Genevieve D, Gatinois V, Lacombe D, Michaud V, Rossi M, Lebreton C, Leheup B, Vincent‐Delorme C, Delahaye A, Van Maldergem L, Cacciagli P, Scheffer H, Saugier‐Weber P, Goldenburg A, Philip N. 2015. KBG syndrome: A series of 20 French patients. Eur Soc Hum Genet Abstract PS11 79 223.
-
- Carey JC, Cohen MM, Jr , Curry CJR, Devriendt K, Holmes LB, Verloes A. 2009. Elements of morphology: Standard terminology for the lips, mouth, and oral region. Am J Med Genet Part A 149A:77–92. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
