

Validity of using ad hoc methods to analyze secondary traits in case-control association studies
- PMID: 27670932
- PMCID: PMC5170877
- DOI: 10.1002/gepi.21994
Validity of using ad hoc methods to analyze secondary traits in case-control association studies
Abstract
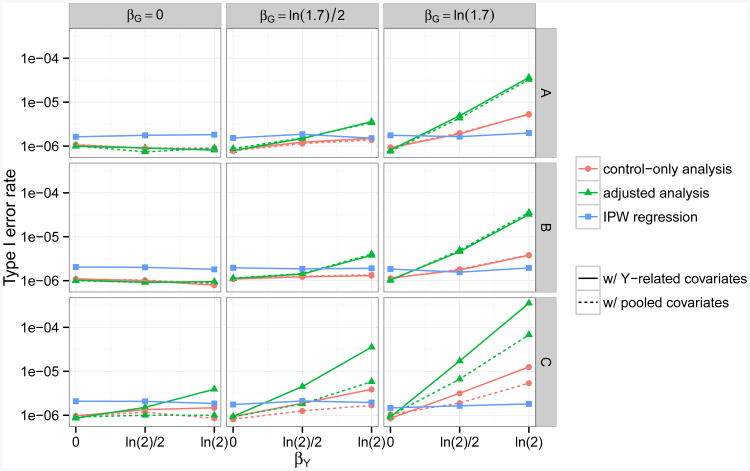
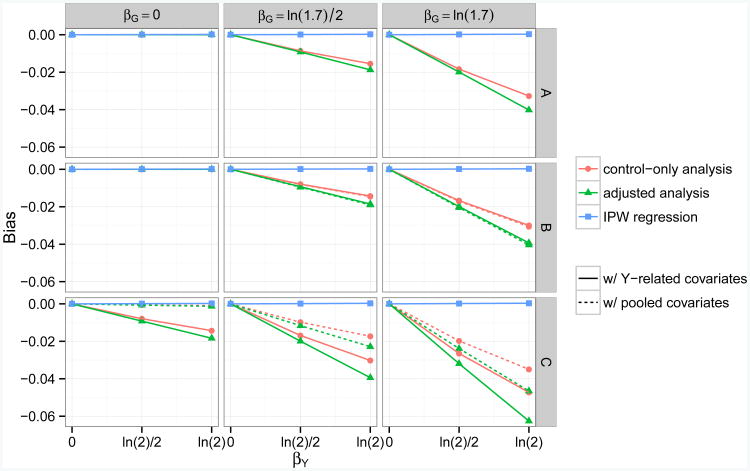
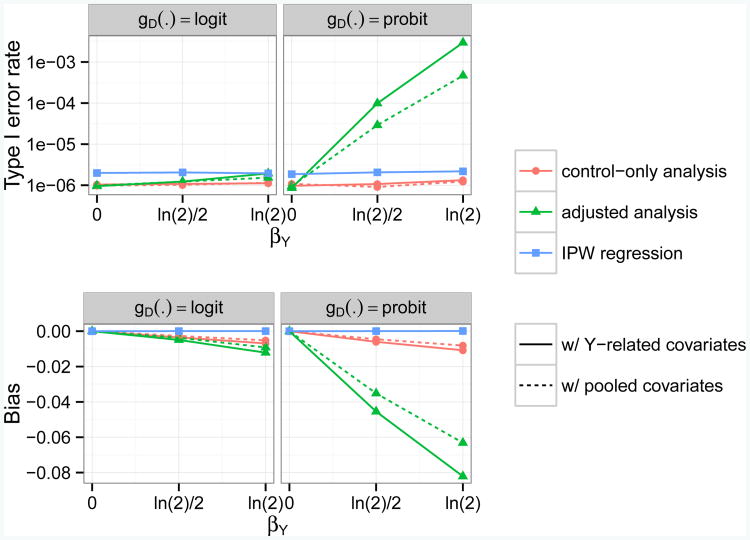
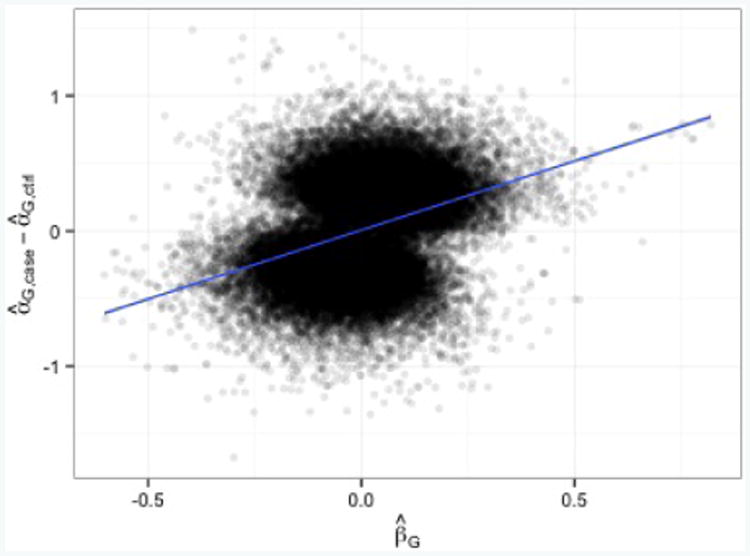
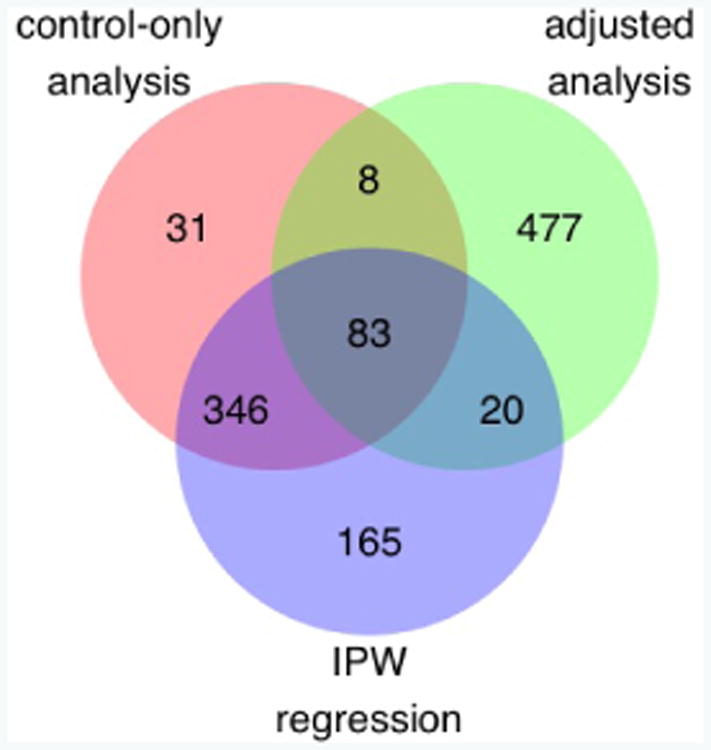
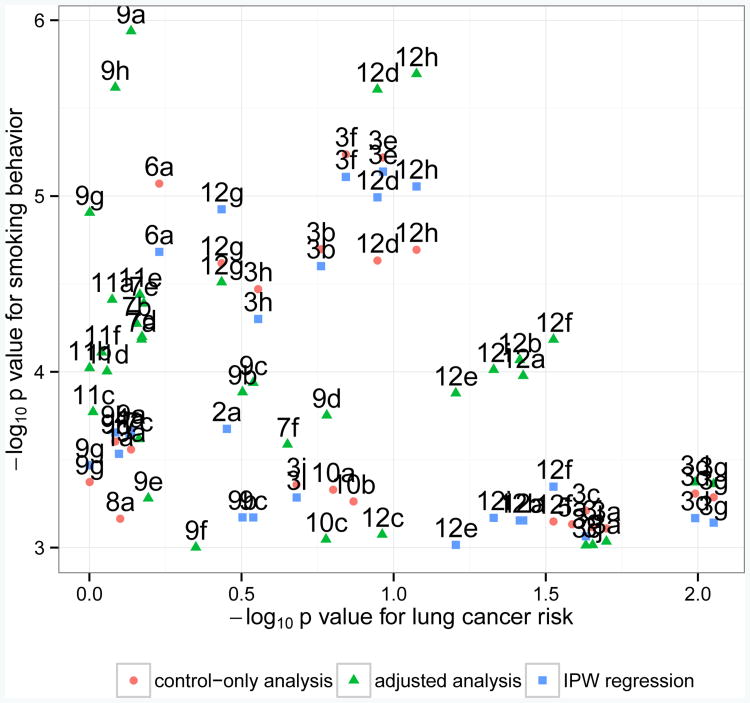
Case-control association studies often collect from their subjects information on secondary phenotypes. Reusing the data and studying the association between genes and secondary phenotypes provide an attractive and cost-effective approach that can lead to discovery of new genetic associations. A number of approaches have been proposed, including simple and computationally efficient ad hoc methods that ignore ascertainment or stratify on case-control status. Justification for these approaches relies on the assumption of no covariates and the correct specification of the primary disease model as a logistic model. Both might not be true in practice, for example, in the presence of population stratification or the primary disease model following a probit model. In this paper, we investigate the validity of ad hoc methods in the presence of covariates and possible disease model misspecification. We show that in taking an ad hoc approach, it may be desirable to include covariates that affect the primary disease in the secondary phenotype model, even though these covariates are not necessarily associated with the secondary phenotype. We also show that when the disease is rare, ad hoc methods can lead to severely biased estimation and inference if the true disease model follows a probit model instead of a logistic model. Our results are justified theoretically and via simulations. Applied to real data analysis of genetic associations with cigarette smoking, ad hoc methods collectively identified as highly significant (P<10-5) single nucleotide polymorphisms from over 10 genes, genes that were identified in previous studies of smoking cessation.
Keywords: SNPs; case-control sampling; genome-wide association studies; linear regression; logistic regression; secondary phenotypes.
© 2016 WILEY PERIODICALS, INC.
Figures
References
-
- Amemiya T. Qualitative response models: a survey. J Econ Lit. 1981;19:1483–1536.
-
- Jiang Y, Scott AJ, Wild CJ. Secondary analysis of case-control data. Stat Med. 2006;25:1323–1339. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
