

SARM1-specific motifs in the TIR domain enable NAD+ loss and regulate injury-induced SARM1 activation
- PMID: 27671644
- PMCID: PMC5068253
- DOI: 10.1073/pnas.1601506113
SARM1-specific motifs in the TIR domain enable NAD+ loss and regulate injury-induced SARM1 activation
Abstract
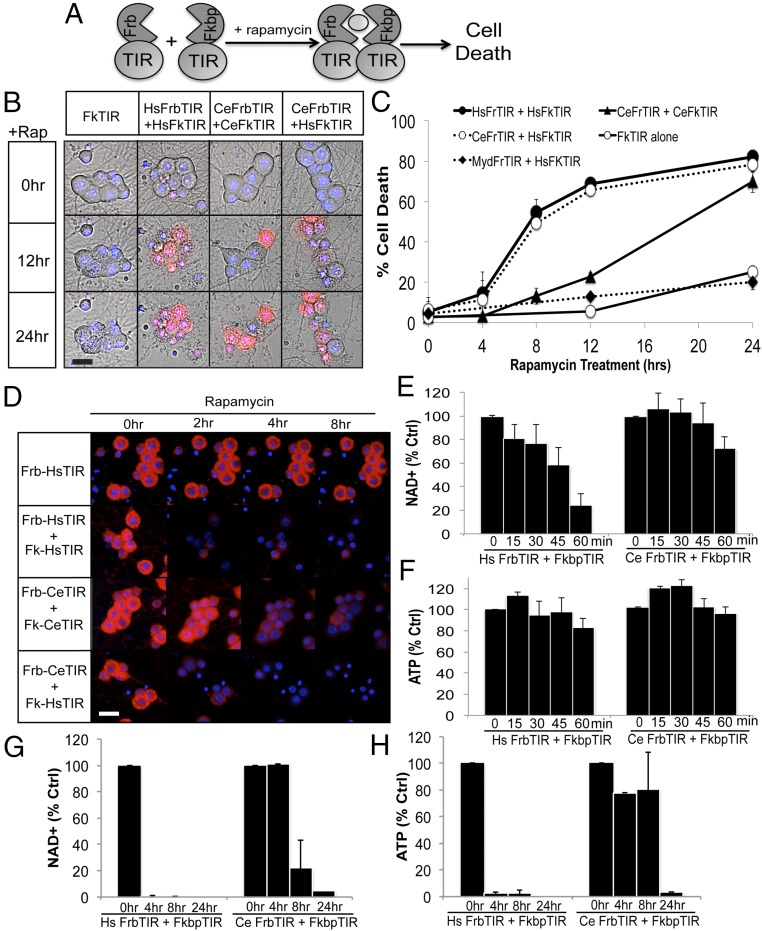
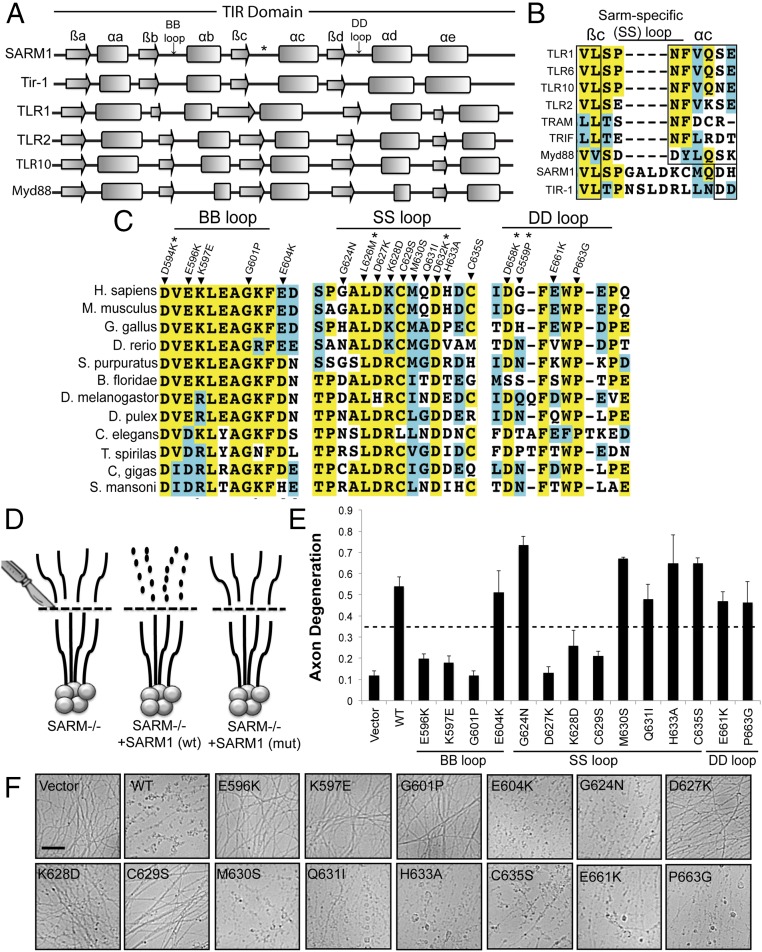
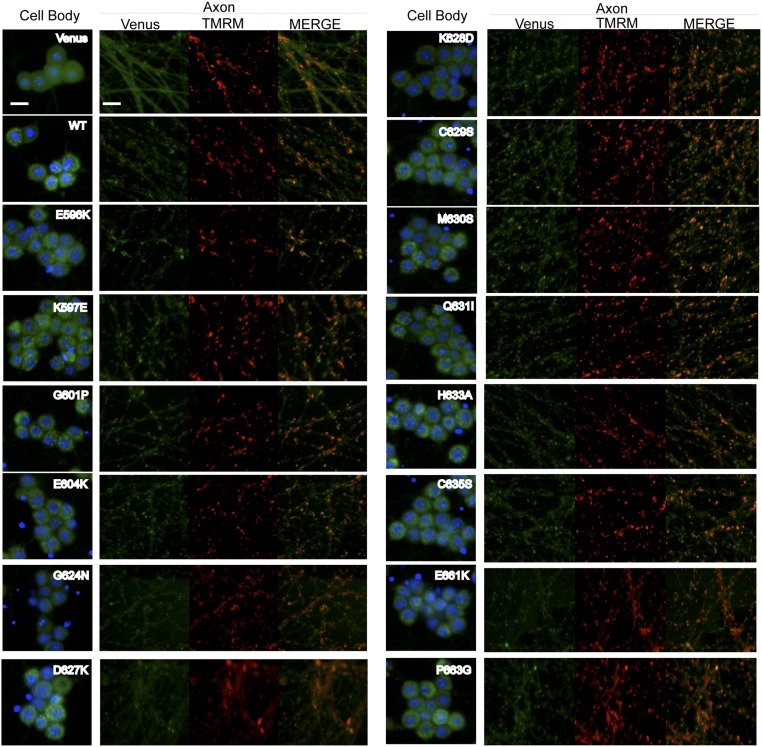
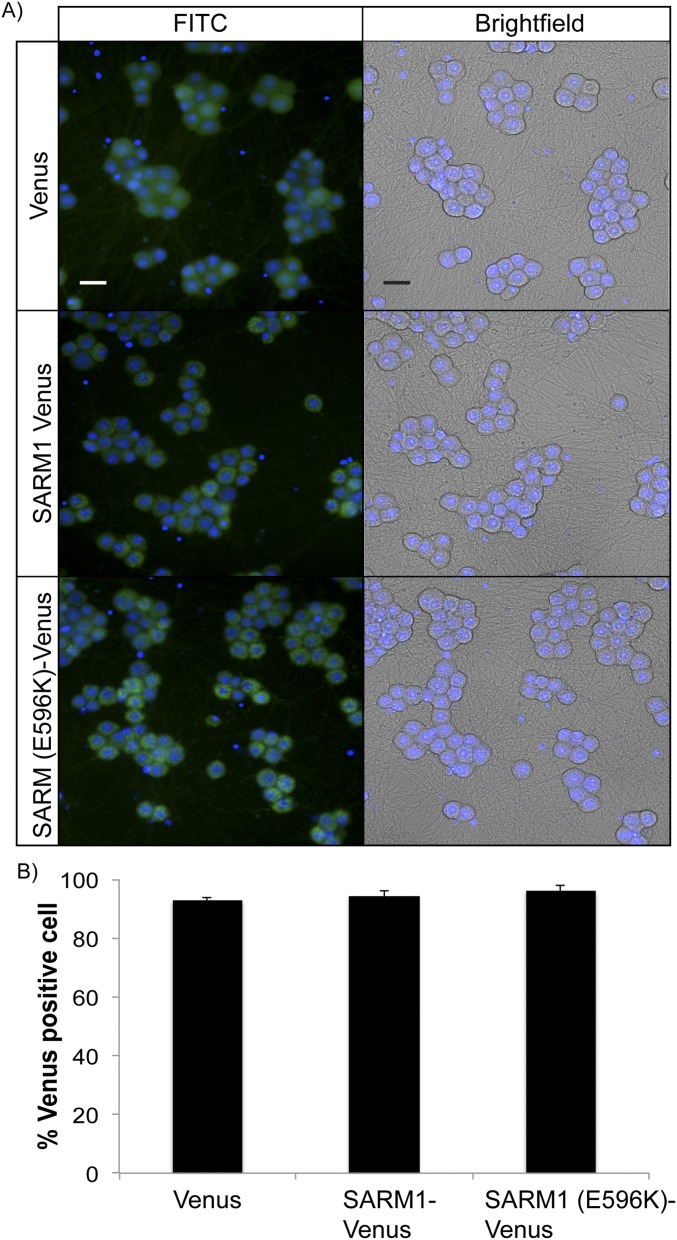
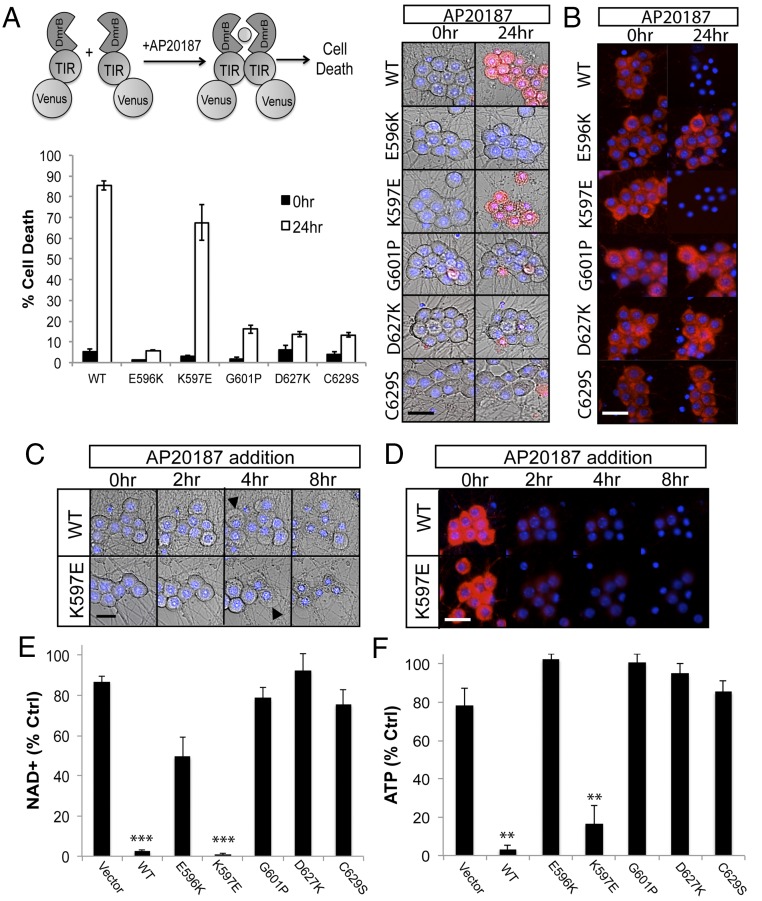
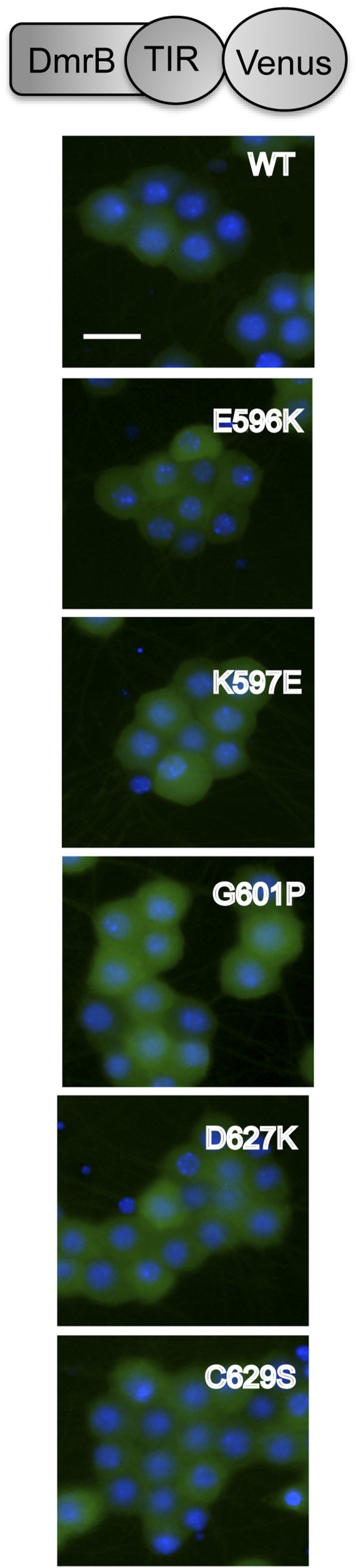
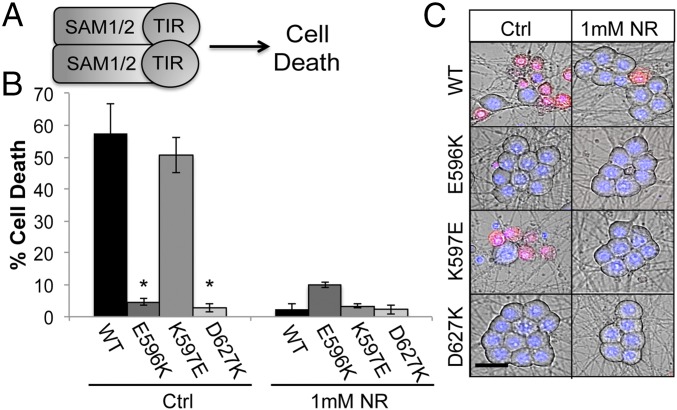
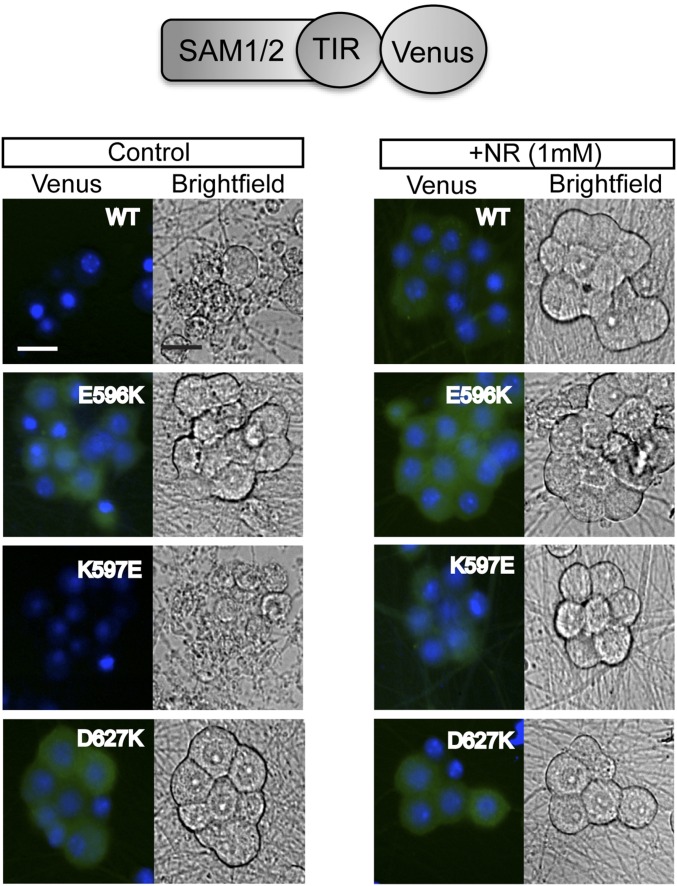
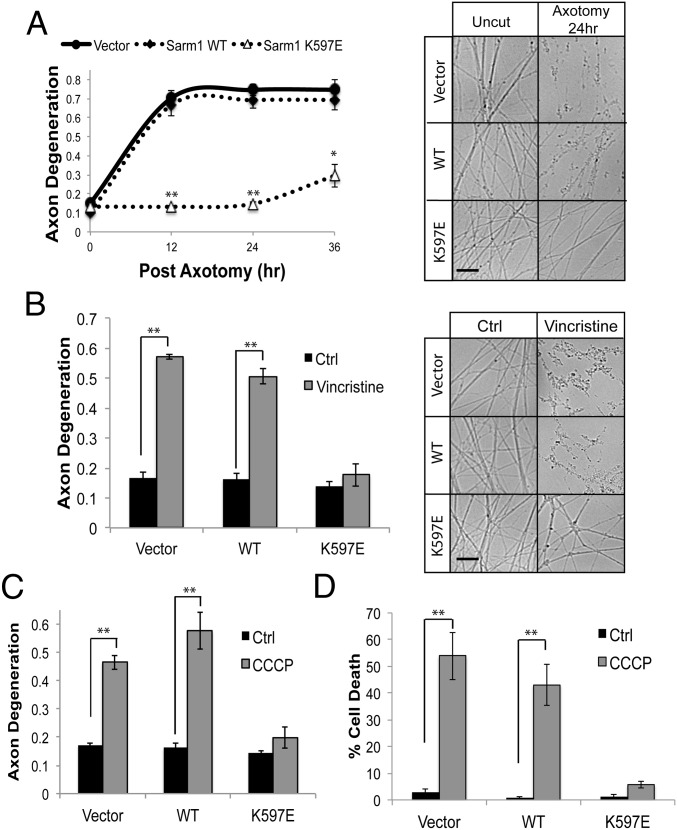
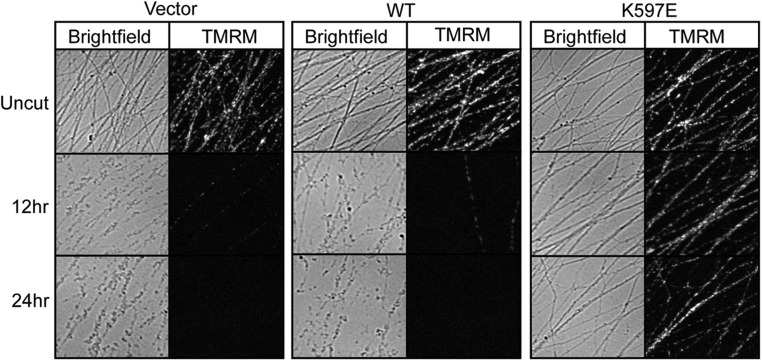
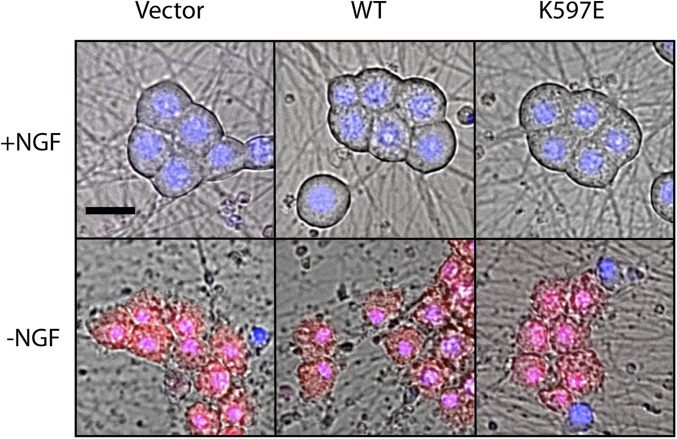
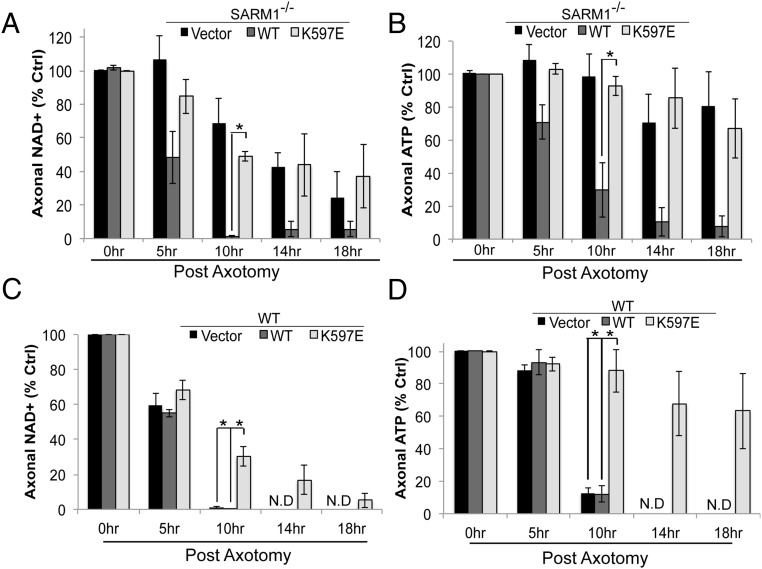
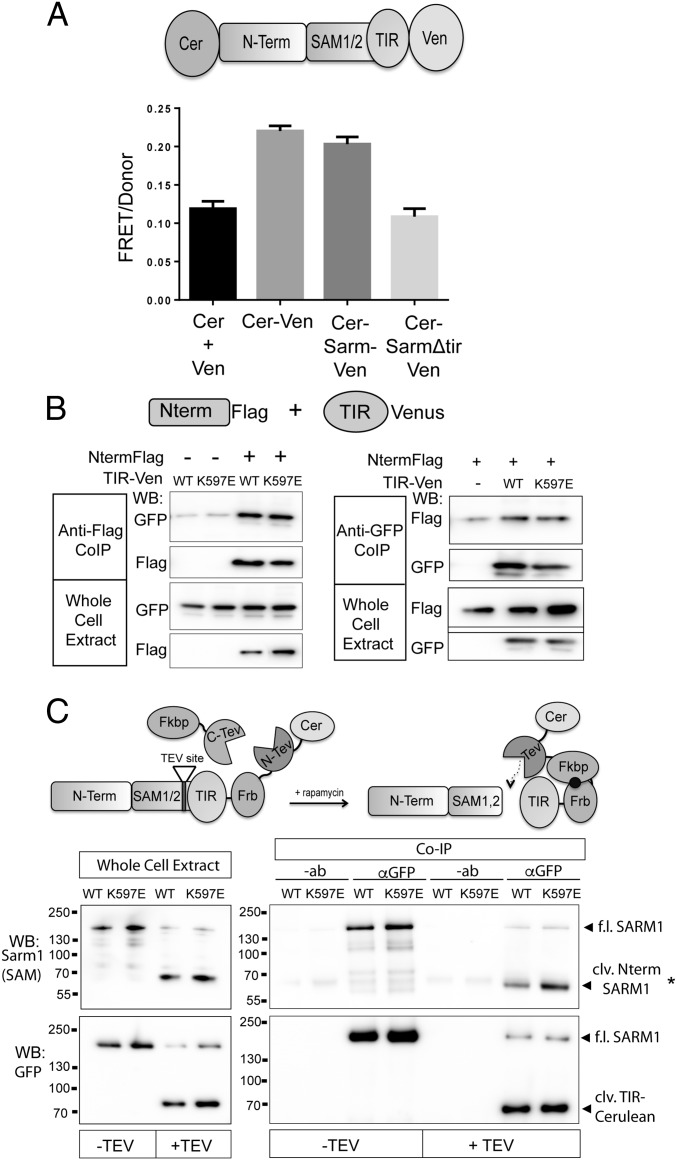
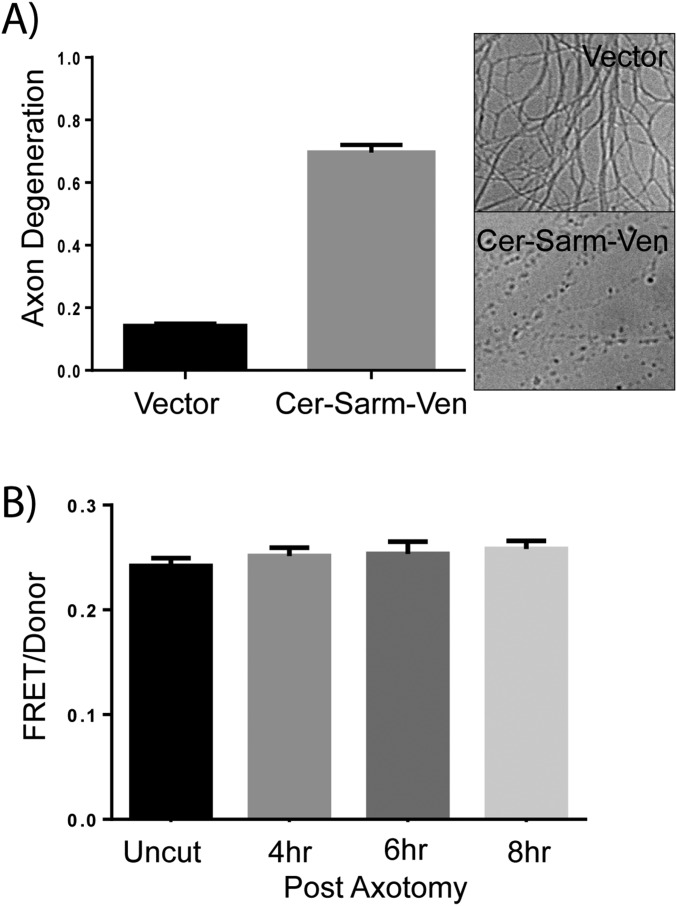
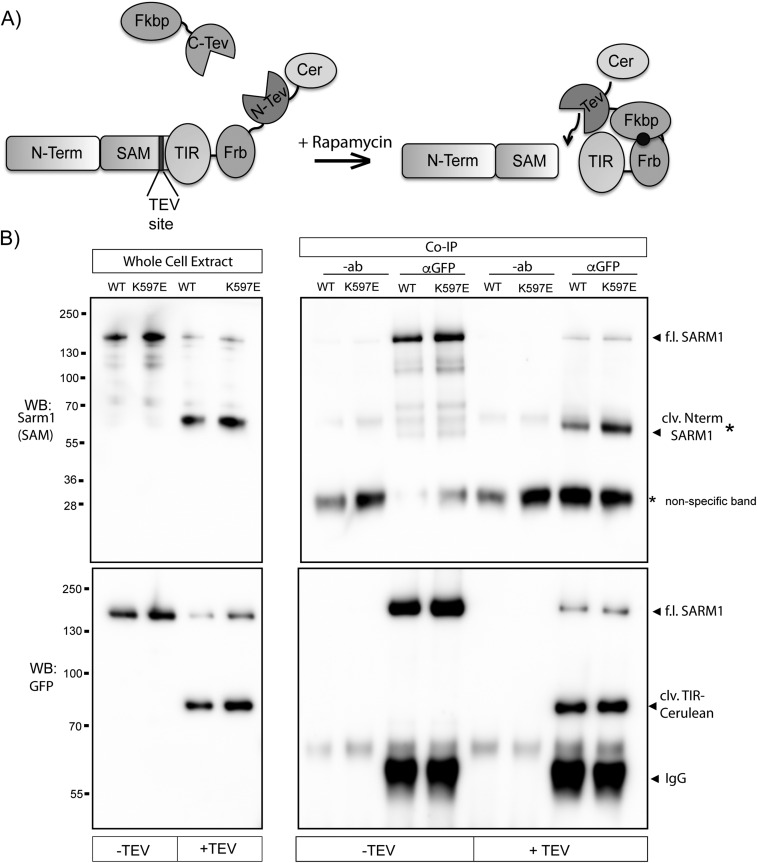
Axon injury in response to trauma or disease stimulates a self-destruction program that promotes the localized clearance of damaged axon segments. Sterile alpha and Toll/interleukin receptor (TIR) motif-containing protein 1 (SARM1) is an evolutionarily conserved executioner of this degeneration cascade, also known as Wallerian degeneration; however, the mechanism of SARM1-dependent neuronal destruction is still obscure. SARM1 possesses a TIR domain that is necessary for SARM1 activity. In other proteins, dimerized TIR domains serve as scaffolds for innate immune signaling. In contrast, dimerization of the SARM1 TIR domain promotes consumption of the essential metabolite NAD+ and induces neuronal destruction. This activity is unique to the SARM1 TIR domain, yet the structural elements that enable this activity are unknown. In this study, we identify fundamental properties of the SARM1 TIR domain that promote NAD+ loss and axon degeneration. Dimerization of the TIR domain from the Caenorhabditis elegans SARM1 ortholog TIR-1 leads to NAD+ loss and neuronal death, indicating these activities are an evolutionarily conserved feature of SARM1 function. Detailed analysis of sequence homology identifies canonical TIR motifs as well as a SARM1-specific (SS) loop that are required for NAD+ loss and axon degeneration. Furthermore, we identify a residue in the SARM1 BB loop that is dispensable for TIR activity yet required for injury-induced activation of full-length SARM1, suggesting that SARM1 function requires multidomain interactions. Indeed, we identify a physical interaction between the autoinhibitory N terminus and the TIR domain of SARM1, revealing a previously unrecognized direct connection between these domains that we propose mediates autoinhibition and activation upon injury.
Keywords: NAD; SARM; axon degeneration; cell death; sarmoptosis.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Multiple domain interfaces mediate SARM1 autoinhibition.Proc Natl Acad Sci U S A. 2021 Jan 26;118(4):e2023151118. doi: 10.1073/pnas.2023151118. Proc Natl Acad Sci U S A. 2021. PMID: 33468661 Free PMC article.
-
SARM1 activation triggers axon degeneration locally via NAD⁺ destruction.Science. 2015 Apr 24;348(6233):453-7. doi: 10.1126/science.1258366. Epub 2015 Apr 23. Science. 2015. PMID: 25908823 Free PMC article.
-
Sarm1-mediated axon degeneration requires both SAM and TIR interactions.J Neurosci. 2013 Aug 14;33(33):13569-80. doi: 10.1523/JNEUROSCI.1197-13.2013. J Neurosci. 2013. PMID: 23946415 Free PMC article.
-
The chemical biology of NAD+ regulation in axon degeneration.Curr Opin Chem Biol. 2022 Aug;69:102176. doi: 10.1016/j.cbpa.2022.102176. Epub 2022 Jul 1. Curr Opin Chem Biol. 2022. PMID: 35780654 Free PMC article. Review.
-
The SARM1 TIR NADase: Mechanistic Similarities to Bacterial Phage Defense and Toxin-Antitoxin Systems.Front Immunol. 2021 Sep 23;12:752898. doi: 10.3389/fimmu.2021.752898. eCollection 2021. Front Immunol. 2021. PMID: 34630431 Free PMC article. Review.
Cited by
-
Emergence of SARM1 as a Potential Therapeutic Target for Wallerian-type Diseases.Cell Chem Biol. 2020 Jan 16;27(1):1-13. doi: 10.1016/j.chembiol.2019.11.002. Epub 2019 Nov 21. Cell Chem Biol. 2020. PMID: 31761689 Free PMC article. Review.
-
Divergent signaling requirements of dSARM in injury-induced degeneration and developmental glial phagocytosis.PLoS Genet. 2022 Jun 23;18(6):e1010257. doi: 10.1371/journal.pgen.1010257. eCollection 2022 Jun. PLoS Genet. 2022. PMID: 35737721 Free PMC article.
-
Neurotoxins subvert the allosteric activation mechanism of SARM1 to induce neuronal loss.Cell Rep. 2021 Oct 19;37(3):109872. doi: 10.1016/j.celrep.2021.109872. Cell Rep. 2021. PMID: 34686345 Free PMC article.
-
Axon degeneration: mechanistic insights lead to therapeutic opportunities for the prevention and treatment of peripheral neuropathy.Pain. 2019 May;160 Suppl 1(Suppl 1):S17-S22. doi: 10.1097/j.pain.0000000000001528. Pain. 2019. PMID: 31008845 Free PMC article. Review.
-
The SARM1 axon degeneration pathway: control of the NAD+ metabolome regulates axon survival in health and disease.Curr Opin Neurobiol. 2020 Aug;63:59-66. doi: 10.1016/j.conb.2020.02.012. Epub 2020 Apr 17. Curr Opin Neurobiol. 2020. PMID: 32311648 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
