

Adolescent Alcohol Exposure Persistently Impacts Adult Neurobiology and Behavior
- PMID: 27677720
- PMCID: PMC5050442
- DOI: 10.1124/pr.115.012138
Adolescent Alcohol Exposure Persistently Impacts Adult Neurobiology and Behavior
Abstract
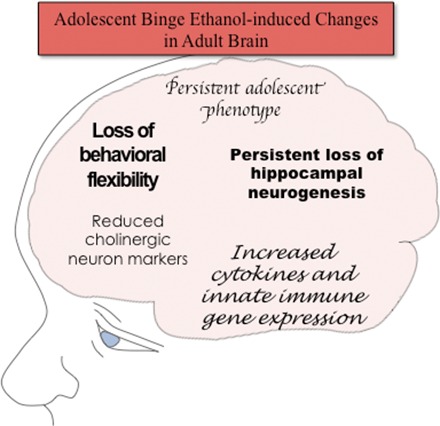
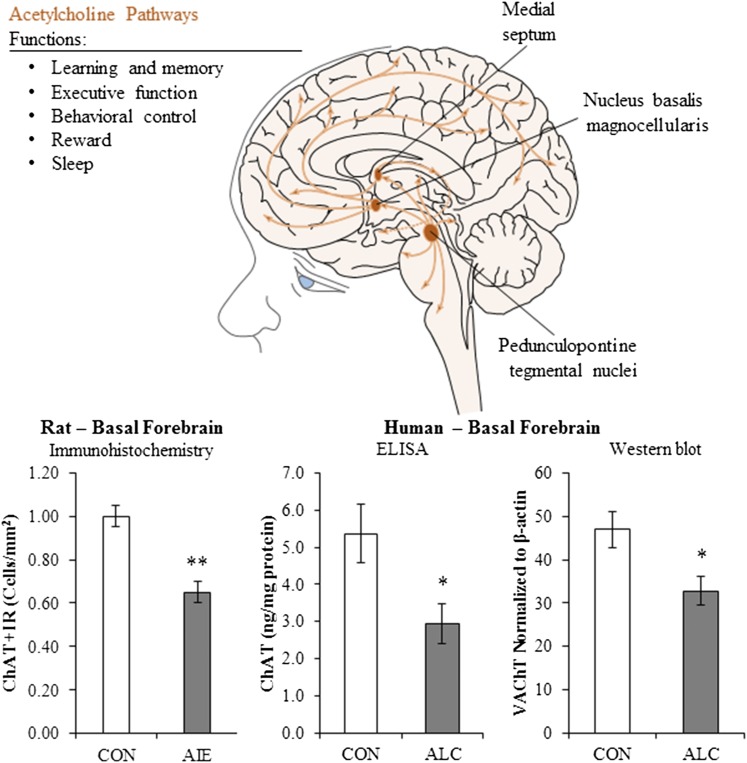
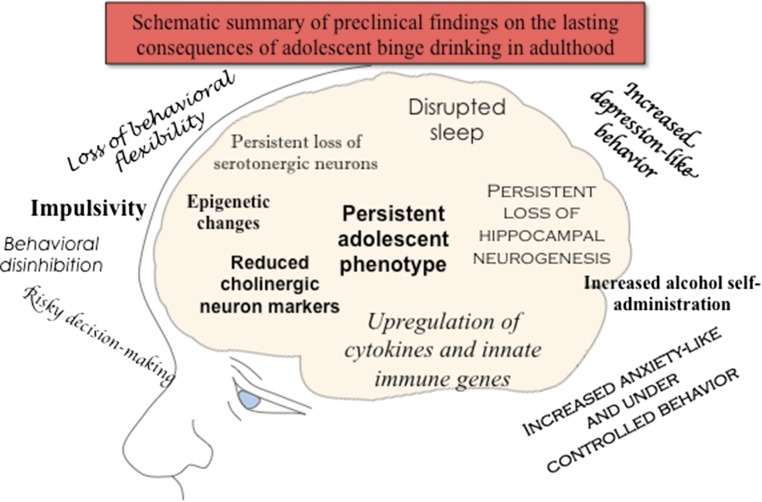
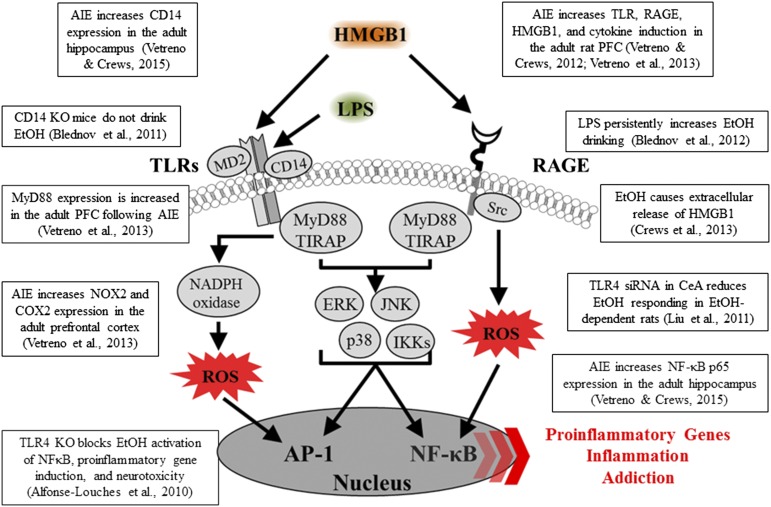
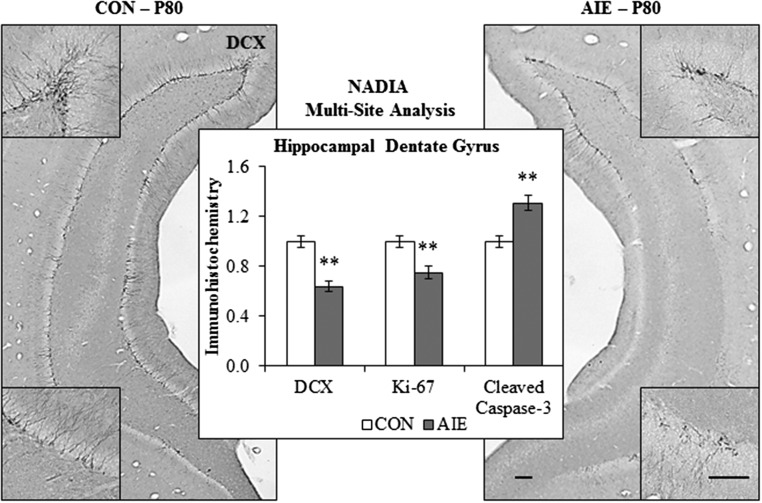
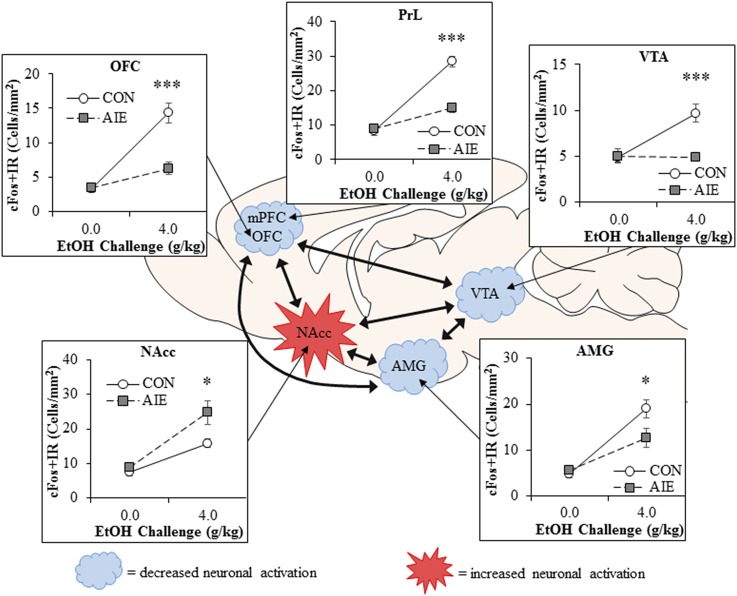
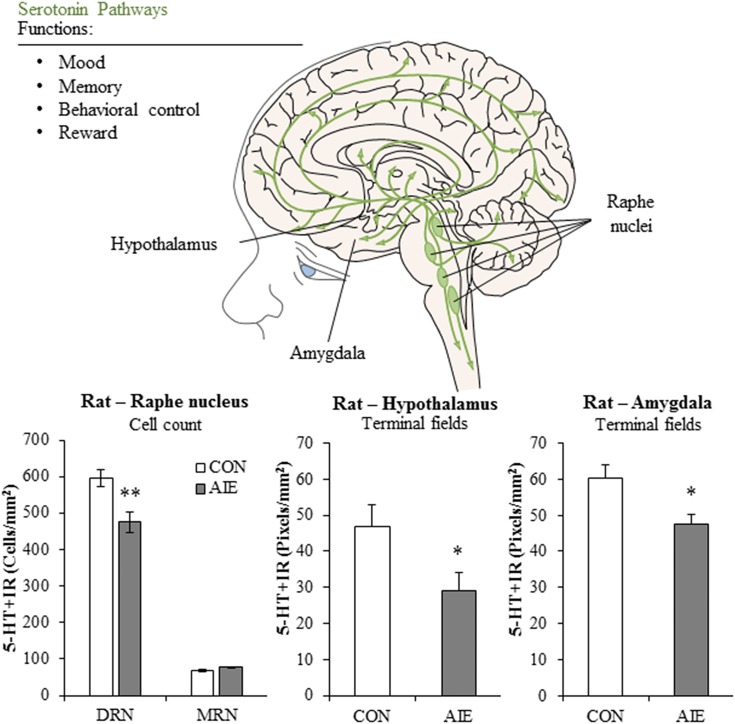
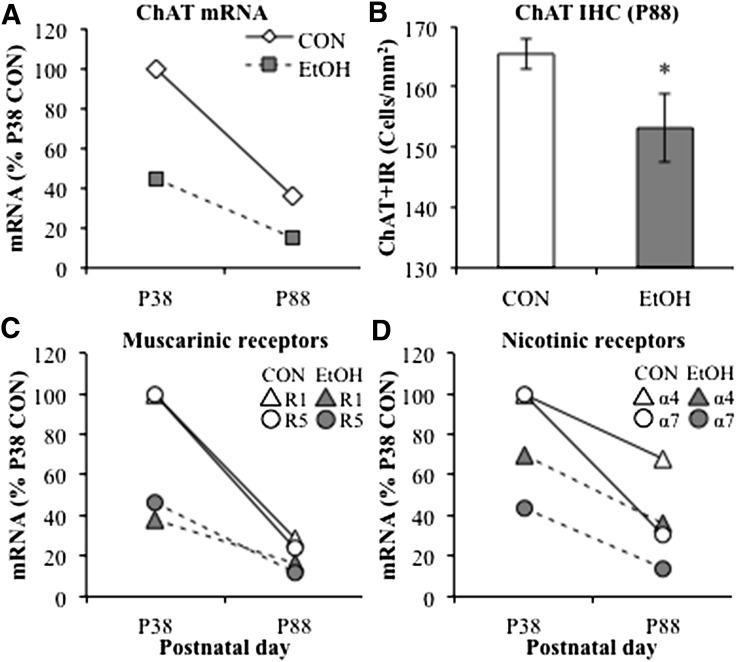
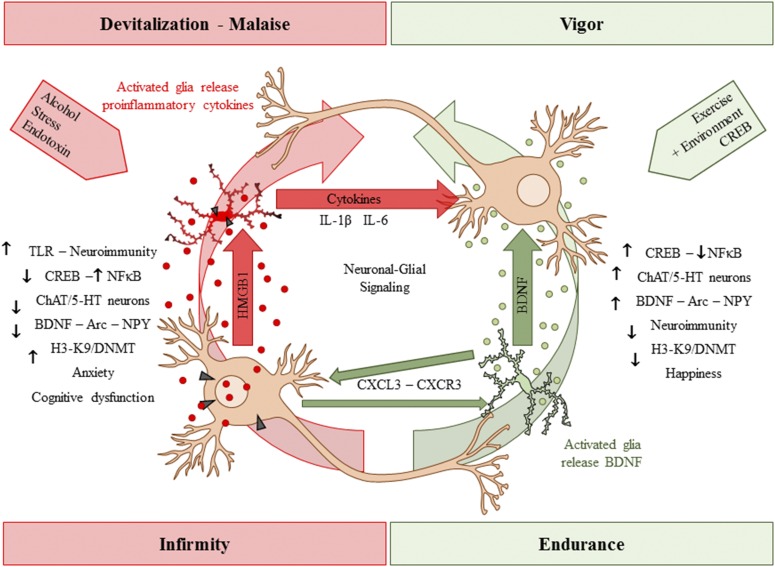
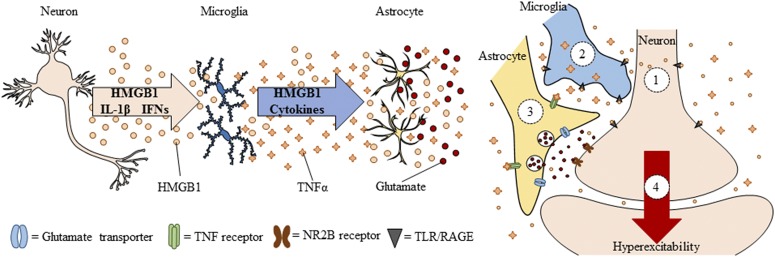
Adolescence is a developmental period when physical and cognitive abilities are optimized, when social skills are consolidated, and when sexuality, adolescent behaviors, and frontal cortical functions mature to adult levels. Adolescents also have unique responses to alcohol compared with adults, being less sensitive to ethanol sedative-motor responses that most likely contribute to binge drinking and blackouts. Population studies find that an early age of drinking onset correlates with increased lifetime risks for the development of alcohol dependence, violence, and injuries. Brain synapses, myelination, and neural circuits mature in adolescence to adult levels in parallel with increased reflection on the consequence of actions and reduced impulsivity and thrill seeking. Alcohol binge drinking could alter human development, but variations in genetics, peer groups, family structure, early life experiences, and the emergence of psychopathology in humans confound studies. As adolescence is common to mammalian species, preclinical models of binge drinking provide insight into the direct impact of alcohol on adolescent development. This review relates human findings to basic science studies, particularly the preclinical studies of the Neurobiology of Adolescent Drinking in Adulthood (NADIA) Consortium. These studies focus on persistent adult changes in neurobiology and behavior following adolescent intermittent ethanol (AIE), a model of underage drinking. NADIA studies and others find that AIE results in the following: increases in adult alcohol drinking, disinhibition, and social anxiety; altered adult synapses, cognition, and sleep; reduced adult neurogenesis, cholinergic, and serotonergic neurons; and increased neuroimmune gene expression and epigenetic modifiers of gene expression. Many of these effects are specific to adolescents and not found in parallel adult studies. AIE can cause a persistence of adolescent-like synaptic physiology, behavior, and sensitivity to alcohol into adulthood. Together, these findings support the hypothesis that adolescent binge drinking leads to long-lasting changes in the adult brain that increase risks of adult psychopathology, particularly for alcohol dependence.
Copyright © 2016 by The Author(s).
Figures
References
-
- Abrous DN, Koehl M, Le Moal M. (2005) Adult neurogenesis: from precursors to network and physiology. Physiol Rev 85:523–569. - PubMed
-
- Acheson SK, Stein RM, Swartzwelder HS. (1998) Impairment of semantic and figural memory by acute ethanol: age-dependent effects. Alcohol Clin Exp Res 22:1437–1442. - PubMed
-
- Alaux-Cantin S, Warnault V, Legastelois R, Botia B, Pierrefiche O, Vilpoux C, Naassila M. (2013) Alcohol intoxications during adolescence increase motivation for alcohol in adult rats and induce neuroadaptations in the nucleus accumbens. Neuropharmacology 67:521–531. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
